





摘要
间歇性缺氧,梗阻性睡眠呼吸暂停(OSA)的主要刺激,诱导炎症,导致早期动脉粥样硬化。环氧氧酶(COX)途径是否有助于间歇性缺氧诱导的动脉粥样硬化仍有待确定。
我们研究了8周间歇性缺氧暴露对COX-pathway基因表达和动脉粥样硬化的影响,以及SC-560抑制COX-1对载脂蛋白E主动脉粥样硬化进展的影响-/-老鼠。尿11-脱氢噻吩焦烷B.2(11-DTXB2)在50个OSA受试者中评估,无心血管危险因子与年龄和体重指数匹配,25种对照,和56个OSA,具有心血管危险因素。
间歇性缺氧显著增加动脉粥样硬化病变的大小,COX-1和血栓素合成酶(TXBS)的mRNA水平。病变大小与COX-1 (r = 0.654, p = 0.0003)和TXBS (r = 0.693, p<0.0001) mRNA水平相关。COX-1抑制仅能减少间歇性缺氧小鼠的病变进展(p = 0.04)。尿11-DTXB2在无心血管危险因素的OSA受试者和对照组中,两者的差异相似,但与无心血管危险因素的OSA受试者相比,有心血管危险因素的OSA受试者增加了13% (p = 0.007)。
尽管阻塞性睡眠呼吸暂停本身与尿11-dTXB增加无关2在间歇性缺氧暴露小鼠和具有心血管危险因素的OSA受试者中,COX-1通路被激活,可能参与了间歇性缺氧诱导的动脉粥样硬化发生。COX-1抑制在预防OSA心血管疾病方面具有临床意义。
介绍
阻塞性睡眠呼吸暂停(OSA)的特点是在睡眠期间部分或全部上呼吸道阻塞反复发作,导致慢性间歇性缺氧(CIH)。这一特征是参与OSA心血管重塑的主要因素[1].在OSA患者中,动脉粥样硬化的早期体征与缺氧严重程度相关[2]但即使在调整混淆因素后。OSA与心血管发病率和死亡率增加有关,并被识别为独立的心血管危险因素(CVRF)[3.].此外,载脂蛋白e缺陷(ApoE)暴露-/-) 老鼠 [4.那5.]和c57bl / 6j小鼠[6.]加速动脉粥样硬化的进展。
动脉粥样硬化是一种慢性炎症性疾病。在许多参与动脉粥样硬化发生的炎症介质中,我们之前证明了花生四烯酸衍生代谢物在OSA患者中的作用。值得注意的是,OSA患者的白三烯B水平升高4.[7.那8.[Cysteinyl-Leukotrienes [9.与血管重塑有关。然而,环氧化酶(COX)依赖的花生四烯酸代谢途径在OSA患者中的研究较少,其在cih诱导的动脉粥样硬化发生中的意义尚不清楚。
血栓素一个2(酸2)和前列环素(PGI2)是花生酸代谢的两个Cox衍生的代谢物。TXA2主要由血小板通过COX 1亚型(COX-1)和血栓素合成酶(TXBS)产生。TXA2被迅速代谢到血栓素b2(TXB2)和11-脱氢血栓烷B2(11-DTXB2),两种代谢物分别可在血浆和尿液中定量。TXA2其TP受体上的结合诱导血小板活化,血管收缩,血管平滑肌细胞增殖,增加了粘附分子的表达[10].PGI的产生2主要依赖于内皮细胞COX-2和前列环素合成酶(PGIS)。PGI2迅速代谢为6-酮-前列腺素F1α(血浆中的6-酮- pgf1 α和尿液中的2,3-二甲基-6-酮-前列腺素F1α (PGI-M)。与TXA相比2, PGI2其IP受体上的结合抑制血小板聚集和血管收缩,并减少趋化性和粘附分子的表达[10].因此,酸2和pgi.2有拮抗性的属性。
最近越来越多的证据表明,COX通路在动脉粥样硬化的发病和进展中发挥了重要作用。事实上,COX途径的药理抑制[11那12或遗传缺失[13那14]延缓动脉粥样硬化小鼠模型的发生。此外,尿11-dTXB2/ PGI2-M比率在既往有严重心血管事件(中风和心肌梗死)病史的老年人中增加[15].因为选择性COX-2抑制剂可能对这一比例产生不利影响,并增加心血管风险[16而COX-1抑制的作用则较少受到关注。此外,关于OSA的数据非常有限,而且相互矛盾,显示要么增加了[17或减少的[18]尿比率11-DTXB2/ PGI2- m。因此,本研究的目的是研究ApoE中COX通路的特征-/-研究其与CIH诱导的动脉粥样硬化发生和OSA相关的早期血管重塑的关系。
材料和方法
实验动物研究
雄披风-/-小鼠(14周龄)购自Charles River Laboratories (L’arbresle, France)。所有动物手术都是按照欧洲保护用于实验和其他科学目的的脊椎动物公约(欧洲理事会,欧洲条约ETS 123,斯特拉斯堡,1986年3月18日)和实验室动物护理和使用指南(NIH出版物编号:no. 1)进行的。85 - 23, 1996年修订)。
在第一组实验中,小鼠被随机分配到8周的CIH(循环21-25%吸气氧分数(FIO2), 60 s循环8 h·day-1)如前所述(每组中的n = 15)或常见的空气)[1].在第二系列实验中,将小鼠暴露于CIH或常氧空气的8周(每组N = 20),并随机接收安慰剂或选择性COX-1抑制剂SC-560(Interchim,Montluçon,法国)(15毫克·kg-1每日(12),在最后4周的暴露中。所有老鼠都被喂食正常的食物,随意在所有的实验中。
暴露结束时,在麻醉下采集血液(氯胺酮/二甲苯100 mg·kg)-1/ 10毫克公斤-1通过腹膜内注射)用于血细胞比容和脂质测量。收获整个主动脉。将腹部主动脉置于RNALATER(Life Technologies,Villebon-Sur-Yvette,France)中,在液氮中冷冻并储存在-80°C直至分析。在将它们的取样后立即置于Tyrode溶液(137mm NaCl,2.7mm Kcl,0.41mm NaHPO的胸膜溶液中4., 2 mM CaCl2, 5 μM MgCl2,11.9毫米Nahco3.用于前列腺分泌测量的5.5毫米葡萄糖)。
动脉粥样硬化病变尺寸量化
如前所述,采用Oil-Red-O (Sigma Aldrich, Saint Quentin-Fallavier, France)染色对主动脉根动脉粥样硬化病变进行研究[5.].对于每个主动脉,使用计算机图像分析(NisElement;尼康仪器公司,梅尔维尔,纽约,美国)。
前列腺的主动脉分泌
胸主动脉在37℃的Tyrode溶液中孵育15分钟,95% O2和5%的co2,在10个钙的Ionophore A23187(Sigma Aldrich,Saint Quentin Ralkavier,法国)的存在下-6M.上清立即在-80°C冷冻以测量前列腺素,干燥主动脉以测量干组织重量。
Cox-途径基因表达
如前所述,使用RNeasy试剂盒(Qiagen, Hilden, Germany)从主动脉分离总mRNA [19]根据制造商的说明,使用随机六甲烷的上标II(Invitrogen,Carlsbad,Ca,USA)使用上标II(Invitrogen,Carlsbad,Ca)反转。使用底漆/探针对进行定量Taqman PCR,使用具有测定的按需(Applied Biosystems; Life Technologies SAS,Saint Aubin,France)(在线补充表S1)。将数据归一化至18s核糖体蛋白mRNA并表示为2——ΔCT.
临床研究
根据赫尔辛基宣言,当地伦理委员会批准了这项研究。所有参与者均给予书面知情同意。
在2007年1月至2011年4月期间,113名新诊断的OSA患者和25名对照组连续进入研究。患者被转介到格勒诺布尔大学医院睡眠实验室的症状提示OSA。排除标准为既往脑卒中或心肌梗死史、已知高血压、非甾体类抗炎药、阿司匹林、类固醇、抗糖尿病药、降压药和降脂药治疗。
设计
CIH对11-dTXB的特异性贡献2研究人员评估了25名年龄和体重指数(BMI)匹配的对照组和50名非肥胖OSA患者(IE。两个OSA受试者的一个健康主题)。所有受试者都没有任何已知的CVRF。样品大小为50个OSA患者和25个对照的样品,以足够的方式计算尿11-DTXB中的30%增加2排泄能力至少为90%,显著性水平为5%。这个样本量的计算是基于健康受试者获得的平均11-dTXB浓度的初步数据2647 pg·毫升-1标准偏差为287 pg·mL-1.作为11-DTXB的分散2我们选择了一个混合模型,每两个OSA受试者中就有一个健康者。
CVRFs(包括肥胖(BMI >30 kg·m-2);高血压(临床舒张血压(BP)> 90 mmHg和收缩压增量)> 140mmHg);血脂血症(低密度脂蛋白(LDL)胆固醇>4.13mmol·L-1或总胆固醇>5.16 mmol·l-1高密度脂蛋白胆固醇<1.03 mmol·l-1);抽烟;和代谢综合征(由国际糖尿病联合会的标准定义[20.])关于11-DTXB的尿排泄2在113名OSA患者的整个队列中进行了评估。根据是否存在这些cvrf对患者进行分层。
持续气道正压(CPAP)对尿11-dTXB的影响2对14例无CVRF的OSA患者和21例CVRF(s)持续CPAP的OSA患者进行浓度研究。CPAP依从性定义为每日使用CPAP持续>4小时[9.].
所有受试者都进行了夜间多导睡眠描记术[9.].睡眠呼吸暂停被定义为呼吸暂停/低吞噬症指数(AHI)≥5个事件·h-1呼吸障碍指数(RDI),包括流量限制发作>15事件·h-1[21].
11-DTXB的尿液样本2在夜间多瘤记录末端07:00 H收集用于生化测量的量化和静脉血液,并在-80°C储存直至以后分析。
如前所述,对97例OSA患者和24例对照组进行颈动脉超声检查[2].
统计分析
使用NCSS97(犹他州凯斯维尔)或SAS (SAS 9.1,美国NC凯里)对混合模型进行统计分析。数据以中位数、第10百分位和第90百分位表示。正态分布采用Kolmogorov-Smirnov非参数检验。用t检验比较常氧空气和CIH小鼠。采用混合模型对年龄和BMI匹配的OSA患者和对照组(2名OSA患者为1名对照组)进行比较。使用Fisher检验对非连续变量进行比较。三组以上的比较采用Kruskal-Wallis检验,随后的两两比较采用Bonferroni多重比较检验。对于接受CPAP治疗的患者,基线值和CPAP后值之间的差异使用配对t检验或Wilcoxon符号秩检验进行分析。用单回归法研究了两个变量之间的关系。将尿液11-dTXB相关变量考虑在内,进行多元线性回归分析2在人类身上。p值<0.05被认为是显著的。
结果
实验动物研究
动脉粥样硬化病变的大小
与常氧小鼠相比,CIH小鼠主动脉根部的动脉粥样硬化病变更高(p = 0.008) (CIH 66 532(32 741-163 224))。相对常氧空气37 675 (6798-75 596)μm2;p = 0.03)。在常氧小鼠中,病灶大小与血浆总胆固醇相关(r = 0.501, p = 0.04),而在CIH小鼠中则不相关。
Cox-途径基因表达
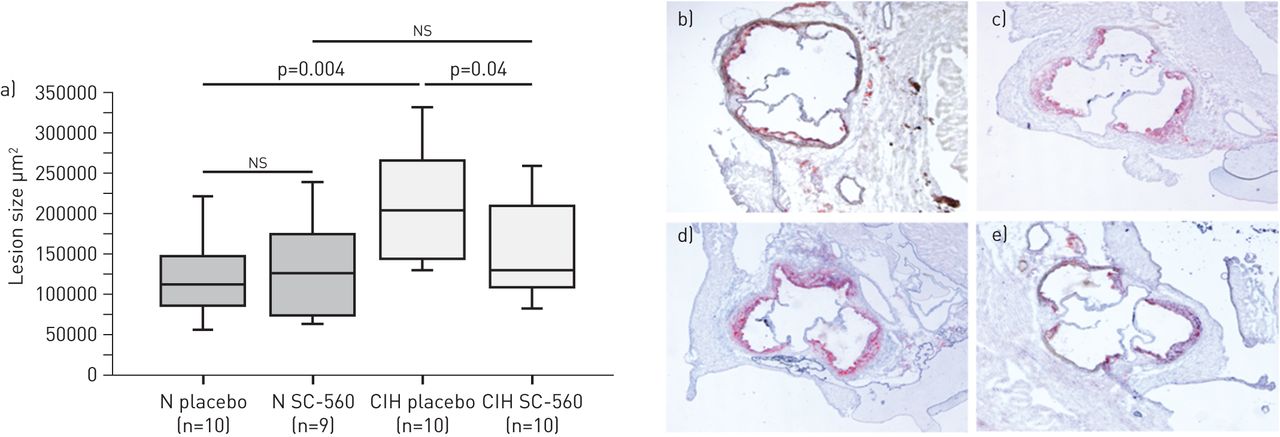
与常氧空气相比,CIH组COX-1和TXBS主动脉mRNA水平显著升高,而COX-2、PGIS、TP和IP受体主动脉mRNA水平在两组间无显著差异(图1一个).COX-1和TXBS的mRNA水平与病变大小明显相关(图1 b).

a)暴露于腹部主动脉慢性间歇性缺氧的小鼠中Cox途径基因的mRNA水平。回归在动脉粥样硬化病变尺寸和B)COX-1和C)血栓素合酶(TXBS)之间的血栓性病变尺寸和主动脉瘤剂量之间。与常氧小鼠相比(控制)相比,数据表示为折叠变化。*:P <0.05相对常氧组。COX-1:环加氧酶1亚型;COX-2: COX 2亚型;txb:血栓素合酶;PGIS:环前列腺素合成酶。
主动脉前列腺素类分泌
a23187刺激6-酮- pgf1 α和TXB2分泌物和6-keto-pgf1α/ txb2CIH小鼠和常氧小鼠的主动脉比例相似(表2).6-keto-PGF1α和TXB2浓度高度相关(r = 0.842,p <0.0001)。
COX-1抑制作用
在常氧空气和CIH组中,SC-560治疗显著降低了TXB2主动脉分泌6-keto-PGF1α相对安慰剂 (表2).总胆固醇和体重的群体之间没有差异(表2).用SC-560治疗在CIH小鼠中显着降低了损伤大小35%,而在常氧气小鼠中没有作用(图2).
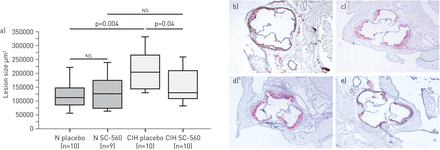
环加氧酶1亚型(COX-1)抑制对慢性间歇性缺氧(CIH)或常氧小鼠动脉粥样硬化的影响(N)。数据以四分位距离(方框)、数据距离(须)和中位数(水平线)表示。ns:不重要的。b)常氧安慰剂,c)常氧SC-560, d) CIH安慰剂和e) CIH SC-560的代表性油-红- o染色照片。
临床研究
11-DTXB.2和阻塞性睡眠呼吸暂停综合症
25对照的基线特征和符合年龄和BMI的50个OSA患者表3.血浆胰岛素,HSCRP,HDL胆固醇,稳态模型评估胰岛素抵抗指数(HOMA-IR),BP,颈动脉内膜介质厚度(IMT)和性别比没有显着差异。正如预期的那样,OSA和对照组之间的多面组参数不同。OSA患者的血浆葡萄糖,总和LDL胆固醇水平显着增加相对控制。尿11-DTXB2与对照组相比无显著性差异(表3).
11-DTXB.2,OSA和心血管危险因素
OSA有或没有CVRFs患者的临床,生物学和多瘤参数表4.具有CVRFS的OSA组具有多乐编程参数,BP,BMI,血浆甘油三酯,总和LDL胆固醇,葡萄糖,胰岛素和HOMA-IR高于OSA组,不含CVRFS(表4).相反,两组在年龄、性别比例、颈动脉IMT、HDL胆固醇和hsCRP (表4).尿11-DTXB2与OSA患者的OSA患者无需CVRFS(图3).在有或没有CVRFs的OSA患者中,11-TXB2轻度至中度患者(AHI <30事件·h)的水平相似-1)和严重OSA (AHI≥30事件·h)-1)(数据未显示)。
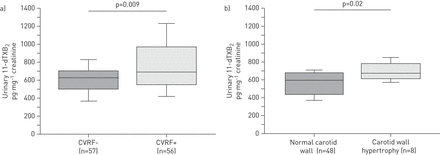
a)尿11-脱氢血栓烷2(11-DTXB2)浓度在阻塞性睡眠呼吸暂停(OSA)患者(CVRF +)和没有(CVRF-)已知的心血管危险因素。b)尿11-dtxb2在有或没有颈动脉壁肥大的OSA患者中的浓度。数据以四分位距离(方框)、数据距离(须)和中位数(水平线)表示。
确定尿11-DTXB增加所涉及的CVRF2,进行简单回归分析。高血压(r = 0.190, p = 0.05)和肥胖(r = 0.242, p = 0.01)与11-dTXB尿排泄量增加有弱相关性2.在多元回归模型中,肥胖仍然是泌尿系11-dTXB的唯一独立预测因素2(r = 0.257, p = 0.04)。
CPAP治疗尿11-dTXB的疗效观察2专注
CPAP治疗至少8周显着降低,RDI和呼吸唤起指数增加,夜间动脉氧饱和度增加(S.敖2)及暗夜S.啊,一个2,并减少了与平均值在一起的时间百分比S.敖2< 90%。CPAP治疗对11-dTXB无影响2OSA的尿液浓度没有或与CVRF(在线补充表S2和S3)。
11-DTXB.2,OSA和血管改造
在无CVRF的OSA患者中,血管肥厚(以颈动脉IMT >0.8 mm定义)与尿11-dTXB增加相关2与没有血管肥大的受试者相比(p = 0.02) (图3 b).
讨论
我们的研究首次证明了Apoe中的Cox途径的激活-/-暴露于CIH的小鼠以及伴有其他cvrf的OSA患者;这种激活与小鼠动脉粥样硬化病变的增加和OSA患者动脉粥样硬化的早期标志物有关。
我们发现,CIH小鼠的动脉粥样硬化病变比常氧小鼠更高,这表明,在我们的模型中,CIH可能加速了动脉粥样硬化的发展。这一结果与前人对载脂蛋白e的研究结果一致-/-小鼠暴露于CIH 2至4周[4.那5.]或在C57BL / 6J小鼠中暴露于CIH 12周[6.].
在本研究中,在常氧空气组和CIH组之间血浆胆固醇水平没有显著差异,在常氧空气组小鼠中,动脉粥样硬化病变的大小与血浆胆固醇相关,而在CIH组中则没有。这些数据表明,在我们的模型中,CIH诱导的动脉粥样硬化可能与脂质紊乱无关,与之前的研究相比,暴露于高脂高胆固醇饮食和CIH同时会加重ApoE的动脉粥样硬化和血脂异常-/-老鼠 [4.]和C57BL/6J小鼠[6.那23].然而,我们最近发现,CIH也通过其他因素发挥促动脉粥样硬化作用,特别是炎症过程[5.].在此后来的假设中,我们的数据显示CIH小鼠中硫醇甲烷途径的激活,因为CIH小鼠的主动脉组织中COX-1和TXBS的mRNA水平增加。此外,这些mRNA水平与动脉粥样硬化病变的相关性与TXA的直接效应一致2在巨噬细胞24].出乎意料的是,主动脉分泌TXB26-酮- pgf1 α,以及TXB2/6-酮- pgf1 α比值在CIH和常氧小鼠中相似。这些结果可能是因为我们测量了A23187刺激主动脉后的前列腺素分泌,而不是基底主动脉的生成。先前在低密度脂蛋白r-敲除(r-KO)小鼠中进行的一项研究表明,高脂肪饮食对动脉粥样硬化发生的加速与tvb基础水平的增加有关26-keto-PGF1α在主动脉弓中的作用[25],但前列腺素的测定采用ELISA法,这不是一种特殊的方法。
我们选择测量TXB26-keto-PGF1α对A23187刺激主动脉,通过我们的分析技术(LC-MS/MS)获得可检测的前列腺素水平,这是高度特异性,但可能不太敏感。然而,在我们的模型中,A23187似乎刺激了TXB2和6-keto-PGF1α的释放水平相同,两者水平高度相关,这可以解释类似的TXB2/ 6-酮-PGF1α与CIH和常氧气小鼠主动脉中的术。我们承认基础TXB的比较26-keto-PGF1α的产生和COX-1和TXBS的Western blot分析可能是关于CIH诱导COX-1和TXBS mRNA水平升高的兴趣所在,而它们的缺失可能代表了我们研究的局限性。
此外,在CIH曝光的最后4周内用选择性COX-1抑制剂SC-560治疗CIH小鼠的动脉粥样硬化进展,为COX途径的CIH依赖性活化提供了进一步的证据。我们已经证明,4周的CIH暴露足以诱导Apoe中的动脉粥样硬化-/-老鼠 [5.[因此,它有兴趣探讨COX-1抑制对已建立的动脉粥样硬化病变的影响。如前所述[12], SC-560治疗对常氧空气小鼠的动脉粥样硬化病变没有影响,尽管通过测量主动脉TXB可以有效抑制COX-1通路2和6-keto-pgf1α分泌物。集体,这些数据在Apoe中表现出来-/-在小鼠中,CIH暴露加速了动脉粥样硬化过程,至少部分通过COX-1途径激活。CIH激活白细胞[26]和OSA患者显示白细胞激活[7.那8.那27].通过调节白细胞、平滑肌细胞和内皮细胞之间的相互作用,TXA2促进和PGI2防止动脉粥样硬化的发生和发展[14].
为了将在CiH小鼠中观察到的硫氨甲烷途径激活,我们测量了11-DTXB的尿排泄2,系统性TXA的经过验证的生物标志物2生产 [10].尿11-DTXB2无CVRF的OSA患者的浓度与年龄和BMI仔细匹配的对照组没有区别,这两个主要的混杂因素经常出现在关注与OSA相关的潜在炎症的研究中。这些数据表明,OSA本身与尿11-dTXB增加无关2CPAP治疗对尿11-dTXB无影响的观察证实了这一假设2的水平。然而,尿11-dTXB增加2先前已在包括肥胖症的心血管疾病患者中描述浓度[28]和高血压[29].与这些数据一致的是,我们发现伴有相关CVRF的OSA患者尿中11-dTXB浓度较高2而非无CVRF的OSA患者此外,在研究的cvrf中,肥胖是尿11-dTXB的唯一独立预测因子2这与之前的一项研究一致,该研究表明尿11-dTXB增加2肥胖女性排泄[28].这一结果提供了肥胖症在OSA患者中肥胖症中肥胖作用的进一步证据,我们以前针对5-脂氧合酶途径描述了[9.].最后,尿11-DTXB2OSA患者颈动脉壁肥大与排泄相关。同样,这些发现与ttxa的增殖作用一致2血管平滑肌细胞[10]但是必须考虑到血管重塑是一种复杂的过程,暗示许多介质,并且Cox途径激活可能不是OSA患者CVRF患者血管重塑的唯一机制。
总之,我们的研究表明ApoE中COX-1通路被激活-/-暴露于CIH的小鼠,这有助于通过该刺激诱导的致动力发生过程的加速。在OSA相关CVRFs的OSA患者中也发现了COX-1途径的这种活化。该研究延伸了前述对白三烯途径描述的疟原酸代购的激活[7.那9.]在OSA中的COX-1途径与血管重塑相关。这些发现开启了新药理学方法的兴趣(双COX和5-脂氧基酶抑制剂的兴趣[30.]),预防OSA患者心血管疾病的发生。
致谢
作者感谢Nathalie Arnold (CHU, Hôpital A. Michallon, Pôle rééducation et physiology, BP217, Grenoble,法国)的统计分析,Jean-François Jourdil, Karine Scalabrino, Cécile Girard (CHU, Hôpital A. Michallon, Laboratoire de Pharmacologie, BP217, Grenoble)和Sandrine Cachot (INSERM, U1042,(格勒诺布尔)寻求专家技术援助。
脚注
这篇文章有补充资料可从www.www.qdcxjkg.com.
支持声明:本研究由美国" le vivier de la recherche médicale "来自格勒诺布尔医学院2010年PHRCResMed基金会《巴黎研究》(Mairie de Paris)French-Swedish基金会和瑞典心脏和肺基金会.
利益冲突:没有宣布。
- 收到了2012年6月18日。
- 公认2012年9月26日。
- ©2013人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}