





抽象
在慢性阻塞性肺疾病(COPD),炎症被认为是从先天免疫的通过危险信号(第一命中)的激活来发起,虽然该机制不容易解释为什么炎症变成慢性。在这里,我们提出了二次打击假说解释了为什么炎症成为COPD患者的慢性。炎症的更严重程度存在于病人谁开发COPD比健康的吸烟者的肺的肺,以及大量的活性氧和活性氮炎性细胞释放的都有可能诱发DNA双链断裂(第二命中)在呼吸道和肺泡细胞,引起细胞凋亡和细胞衰老。DNA损伤反应和衰老相关分泌表型(SASP)也可能被激活,导致产生促炎性细胞因子。这些促炎细胞因子进一步刺激炎症细胞浸润,通过正反馈机制加剧细胞衰老和SASP。这种恶性循环,其特征在于通过相辅相成的炎症和DNA损伤,可能导致在COPD患者中的炎症转为慢性。我们的假设有助于解释为何COPD往往发生于老年人,为什么炎症逐渐加重,为什么炎症甚至戒烟后继续,为什么COPD与肺癌有关。
抽象
恶性循环,涉及相辅相成炎症和DNA损伤,可能导致COPD慢性炎症http://ow.ly/nYW4u
介绍
慢性阻塞性肺疾病(COPD)的炎症目前被理解为包括两个阶段:先天免疫阶段,炎症被危险信号激活(损伤相关分子模式(DAMPs)),以及随后的获得性免疫阶段[1,2]。从先天免疫获得性免疫的免疫反应和后续进展的激活与谁开发COPD症状吸烟者的自然病程,轻度患者病谁开发后来严重的疾病是一致的。然而,这种模式不容易解释为什么患者炎症与COPD变成慢性(与健康的吸烟者的情况),为什么在COPD患者的炎症逐渐加重,或为什么在COPD患者的炎症持续存在,即使他们已经停止吸烟。
由于肺部炎症和肺老化密切相关的,COPD是常常被称为加速或异常肺老化的疾病,或肺的inflammaging的疾病[3-6]。肺老化已建议从损坏的依赖于时间的积累,细胞和大分子[导致3-6],而DNA损伤被认为是一种类型的损害已经在老化牵连[7]。在本文中,我们将介绍的证据支持,其中DNA损伤,如DNA双链断裂,涉及一种机制,通过它发生在COPD炎症可以随后成为慢性的概念。在此基础上的证据,我们建议在炎症二次打击假说由一个“危险信号”(第一击)和DNA损伤(第二次打击)所产生的持续性炎症开始参与COPD的发展。
在COPD中的免疫应答的激活逐步
C奥西奥等。[1]提示慢性阻塞性肺病患者肺部的免疫反应是逐步激活的。在第1步天然免疫激活过程中,一种危险信号,如高迁移率族蛋白盒1蛋白、ATP或尿酸(也称为[8]),由被烟草烟雾破坏的细胞(坏死细胞)释放;这种危险信号可以激活多种受体,包括Toll样受体、晚期糖基化终产物受体、嘌呤能受体和炎症细胞内的受体。这些受体的激活反过来刺激白细胞介素(IL)-1α和IL-1β的产生,从而激活中性粒细胞和巨噬细胞[2]. 步骤2代表从先天免疫到后天免疫的过渡阶段;在此步骤中,CD4+T细胞和CD8+当树突状细胞表达坏死细胞和组织释放的抗原时,t细胞增殖、分化和活化。当第2步免疫耐受不足时,可能会发生向获得性免疫激活的过渡(第3步),导致细胞毒性CD8活性的严重炎症和组织损伤+T细胞,T辅助(TH)1 CD4+T细胞,CD4 Th17细胞+T细胞和B细胞。C奥西奥等。[1]表示,在许多吸烟者处理前进没有比步骤1中,在此期间,先天免疫的激活发生。在谁开发COPD患者,但是,从步骤1中的处理前进到步骤2,从而增加在疾病严重性和获得性免疫的在步骤3 [随后的活化1]。
逐步激活免疫应答的这一理论是在谁开发COPD吸烟者中获得的数据一致,表明一个渐进的病理使病人谁是轻度不适随之成为重症。尽管如此,上述理论并没有解决许多问题。1)为什么COPD在老年人中更常见?2)为什么发生在COPD炎症逐渐恶化?3)为什么类固醇往往是无效的,除非加剧的情况下?4)为什么肺癌往往发展为COPD的并发症?5)为什么在COPD炎症甚至戒烟后坚持?例如,分步激活免疫应答的理论认为作为原因的自身免疫反应的参与,为什么戒烟后炎症持续存在;遗憾的是,迄今为止进行的研究提供了不一致的结果[1,2,9-12]。我们认为,解决上述问题的另一种机制可能依赖于DNA损伤,而不是简单地依赖于免疫反应的逐步激活。具体来说,累积性DNA损伤可以解释慢性阻塞性肺病患者炎症变为慢性的原因,大量积累的数据支持慢性炎症与DNA损伤之间的联系。
慢性炎症诱导活性氧和活性氮对DNA损伤的诱导作用
慢性炎症被广泛公知的通过引起DNA损伤[诱发癌变13]。例如,由感染幽门螺杆菌肝炎病毒和人乳头状瘤病毒分别引起胃癌、肝癌和宫颈癌[14]。即使在没有感染相关原因的慢性炎症病例中,也有报道称癌症发病率很高(例如,与溃疡性结肠炎和克罗恩病相关的结直肠癌,与石棉接触引起的炎症相关的肺癌和间皮瘤,与COPD相关的肺癌)[13,15]. 这些关系的机制包括活性氧(ROS)和活性氮(RNS)的产生,它们可以引起DNA的严重损伤,包括炎症细胞的氧化、硝化和双链断裂[13,16-19]。
由DNA损伤的慢性炎症的诱导
DNA损伤是细胞凋亡和细胞衰老的原因,以及癌发生的一个原因,以及DNA损伤最近已开始引起注意作为慢性炎症的原因[20.-26]。例如,已经作为衰老的DNA双链断裂的结果的细胞产生许多phlogogenic物质(例如IL-1α,IL-6,IL-8,粒细胞 - 巨噬细胞集落刺激因子,生长调节致癌基因α,单核细胞趋化蛋白(MCP)-2,MCP-3,和基质金属蛋白酶1,-2,-3,-12,-13和-14)[21-26]. 衰老相关分泌表型(SASP)被用来描述产生这些金源物质的衰老细胞[27]。ATM、MDC1、NBS1、53BP1、BRCA1、Chk2等被认为是导致这种表型的分子介质;这些介质被激活的DNA双链断裂和传输的DNA损伤反应,它激活核转录因子(NF) -κB [23-26]。NF-κB在早衰的小鼠的抑制已经显示出延缓DNA损伤诱导的炎症,细胞衰老和年龄相关疾病在多个器官[28]。在激活DNA损伤反应时产生的促炎性细胞因子不仅可以激活炎性细胞,还可以通过IL-1、IL-6和IL-8的自分泌和旁分泌作用来增强细胞衰老和SASP [23-26,29]。此外,在活的有机体内动物实验表明,结肠炎变成慢性在自动取款机-/-小鼠,其具有降低的能力修复DNA双链断裂与对照组相比自动取款机+ / +老鼠 [30.]。因此,最近的研究支持了DNA损伤诱导的慢性炎症的概念。
总结上述的研究结果,下面的正反馈回路可以负责的慢性炎症,在COPD的发展:炎性细胞活化→生产的ROS / RNS→DNA双链断裂→活化DNA损伤repsonse的→诱导细胞的衰老→SASP→促炎性细胞因子的产生→炎性细胞的活化[23,24]。通过激活这个正反馈环,形成了一个恶性循环,慢性炎症导致DNA损伤;如果不能修复,这种DNA损伤反过来又会加剧炎症[23,24]。在慢性阻塞性肺病,DNA损伤可从ROS或RNS产生炎性细胞以及ROS存在于烟草烟雾[释放31]。事实上,累积的证据表明DNA损伤的慢性阻塞性肺病发生[32-42]。
DNA损伤和SASP如COPD的炎症介质的持久源
许多研究者,包括我们自己的团队,已经报道了COPD患者肺组织中发生的DNA损伤(包括DNA氧化和甲基化、微卫星DNA不稳定性、杂合性丧失、DNA双链断裂)[32-38]. 外周血淋巴细胞中也观察到DNA断裂[40,43]。本烟草烟雾中ROS和ROS或RNS产生的炎性细胞被认为是慢性阻塞性肺病这种类型的DNA损伤的重要原因[42]。
使用抗γH2AX抗体荧光免疫染色使得有可能检测DNA双链断裂以高灵敏度[44]. 我们最近对人肺组织的研究表明:1)对照组吸烟者肺组织中未观察到ROS介导的DNA双链断裂,2)DNA双链断裂不仅诱导细胞凋亡和细胞衰老,而且诱导慢性炎症通过NF-κB依赖型促炎性细胞因子的产生通过衰老细胞(SASP)38,43,45]。同样,A姆塞勒姆等。[46]结果表明,COPD患者衰老的肺血管内皮细胞产生了IL-6、IL-8、MCP-1和可溶性细胞间粘附分子-1等生金质物质。此外,通过动物实验,我们发现当DNA损伤在小鼠Clara细胞中被特异性地诱导时,Clara细胞的衰老和慢性气道炎症与衰老的Clara细胞产生促炎性细胞因子有关[47]。ÿAO等。[48研究还表明,激活抗衰老sirtuin 1/FOXO3通路可以减轻小鼠的细胞衰老、炎症和肺气肿。总之,这些结果提示DNA损伤、细胞衰老和SASP参与了COPD的发病过程。
慢性阻塞性肺病炎症变为慢性的机制:危险信号加DNA损伤,或称双重打击假说
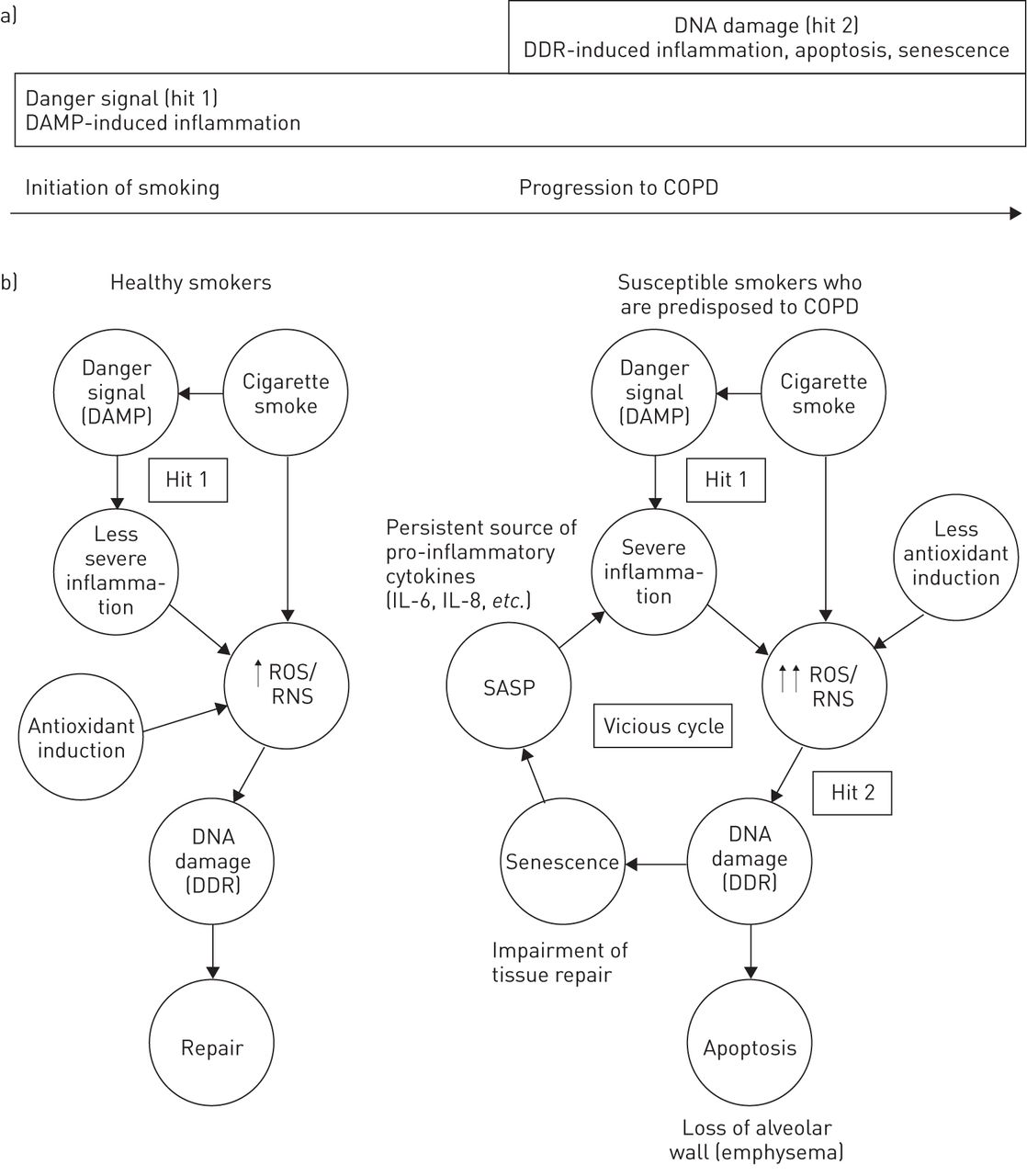
基于上述数据,我们提出了一个解释慢性阻塞性肺病(COPD)患者炎症如何变为慢性炎症的双重假设(图1)。两个命中假说由炎症通过危险信号和炎症的DNA损伤而产生的永久化的起始。如先前由C中所描述奥西奥等。[1,我们认为先天免疫被危险信号激活(第一次攻击)是导致吸烟者炎症的最初诱因。由于一种促进炎症发展的遗传易感性,更严重的炎性细胞浸润发生在“易感”吸烟者的肺中,这些吸烟者比健康吸烟者更容易患慢性阻塞性肺病。由于“易感”吸烟者的抗氧化防御系统活性较低,炎症细胞释放的大量ROS或RNS与烟草烟雾中已经存在的ROS共同诱导气道和肺泡细胞DNA的双链断裂(第二次损伤)。导致气道和肺泡细胞凋亡和细胞衰老,激活DNA损伤反应和SASP,产生促炎细胞因子。这些促炎细胞因子进一步刺激炎性细胞的浸润,形成一个正反馈回路,在这个回路中,细胞衰老通过自分泌和旁分泌作用加强,并促进促炎细胞因子的产生通过该SASP。在COPD患者的炎症被认为是因为炎症和发生由于这个正反馈的结果的DNA损伤之间的恶性循环,成为慢性的。相较于“易”吸烟者,健康吸烟者不发展的DNA损伤[32-35,38这大概是因为导致产生较少量的ROS或RNS,通过抗氧化剂诱导更有效的适应和/或更有效的DNA修复机制的炎性细胞浸润的严重程度较低的],。基于这一假设,我们建议,在吸烟者的DNA损伤是在进展为慢性阻塞性肺病确定事件。换句话说,当基因毒性应激(DNA损伤)加入到烟草烟雾的细胞应激(危险信号)COPD的发展。
二次打击假说,由一个危险信号加上DNA损伤,慢性阻塞性肺疾病(COPD)说明慢性炎症。一)在吸烟,危险信号(命中1)是在肺中的触发启动炎症(C的假说奥西奥等。[1])。根据two-hit假说,有COPD倾向的“易感”吸烟者发生DNA损伤(hit 2),导致细胞凋亡和衰老,并激活DNA损伤反应(DDR)和衰老相关分泌表型(SASP),导致促炎细胞因子的产生。不像健康的吸烟者,“易受影响的”吸烟者会发展成DNA损伤(例如。双链在气道符)和肺泡细胞,大概是因为严格的炎性细胞浸润和较大量的活性氧物质(ROS)/活性氮(RNS)的生产,抗氧化剂的效率较低的诱导,和/或更低效DNA修复机制。这反过来又导致DDR,细胞凋亡,衰老的激活,和SASP和促炎细胞因子。的促炎细胞因子然后加紧炎性细胞浸润,形成一个正反馈环路进一步诱导ROS / RNS生产和DDR,细胞凋亡,衰老的激活,和SASP。以这种方式,一恶性循环炎症和DNA损伤之间建立,且炎症变成慢性。DAMP:损伤相关分子模式;IL:白细胞介素。
Ťzortzaki和Siafakas[42假设树突状细胞将携带基因突变(体细胞突变)的肺泡细胞识别为“非自身”,这些基因突变是由于DNA氧化损伤而产生的,从而导致宿主免疫系统的持续激活;然而,在我们的假设中,我们考虑了DNA损伤反应和SASP的激活通过DNA双链断裂负责慢性炎症的机制。然而,这两个假设并不冲突彼此:它们都涉及到的概念,即DNA损伤导致炎症成为慢性慢性阻塞性肺病。
其他解释慢性阻塞性肺病炎症变为慢性的理论也被提出。例如,自身免疫可能是慢性阻塞性肺病慢性炎症的原因通过其放大正反馈回路的能力,其特征是炎症和自身反应条件的相互强化[1,49]。此外,慢性感染可能不仅是慢性阻塞性肺病变为慢性炎症的原因,也是老年人中慢性阻塞性肺病患病率较高的原因,老年人的免疫监视能力较弱,对不必要的免疫反应的控制也较差[50,51]。
以下的研究来检验二次打击假说解释COPD慢性炎症的有效性。首先,DNA损伤的过程中COPD的早期阶段的实际发生将需要被确认。DNA损伤已经报道COPD患者的肺组织出现,但许多报告的结果都是在研究中获得的,与患者谁了控制相比,吸烟者中度至重度COPD [32-36,38]。如果DNA损伤的发生是区分健康吸烟者和COPD患者的决定性事件,那么即使是轻度COPD患者也应该发生DNA损伤,并且应该随着COPD严重程度的增加而进展。其次,应进行动物实验以确认由于DNA损伤而产生的类copd损伤。虽然我们之前报道过慢性气道炎症是由于小鼠俱乐部细胞的DNA损伤引起的[47]无论DNA损伤是肺气肿的病因还是气道重塑,都应采用长期动物模型进行检测。这类研究的可能动物模型包括长期暴露于烟雾中,或使用DNA修复能力降低的动物,如自动取款机-/-老鼠。第三,是否COPD患者的DNA损伤的发生仅仅是由于DNA损伤的增加,修复能力的下降,还是这两种机制的结合必须加以研究。既往研究报道,ROS抑制DNA失配修复酶活性,COPD患者DNA双链断裂的非同源末端连接修复所需要的蛋白Ku86表达降低,提示COPD患者修复DNA损伤的能力下降[52,53]。第四,除了DNA等遗传变化的双链断裂,诸如组蛋白脱乙酰化,微卫星DNA不稳定,DNA甲基化,端粒缩短和微小RNA的改变,是否参与炎症和DNA损伤之间的恶性循环应检查[6]。
如果DNA损伤的发展是确定事件在吸烟者是否发展COPD,因为在这个假设提出了DNA的存在双链断裂可能作为COPD发展的生物标志物。例如,使用抗γH2AX抗体在从吸烟者在痰或支气管肺泡灌洗液样本,或者在外周血淋巴细胞获得的上皮细胞的DNA双链断裂的检测可能是作为用于治疗COPD的早期检测方法是有用的。此外,防止DNA损伤(如化工,维生素和其他营养物质)化学预防剂可能不仅对预防癌变,也为预防COPD的有用。
慢性阻塞性肺病(COPD)的二击模型与发病机制的相关问题
基于two-hit假说的假设,关于COPD发病机制的问题可以回答如下:1)为什么COPD在老年人中更常见?根据我们的假设,COPD在老年人中可能更常见,因为DNA损伤需要很长时间才能积累。这一概念与突变积累理论是一致的,突变积累理论认为:逐渐积累的DNA损伤在老年时产生有害影响[54]。2)为什么炎症的慢性阻塞性肺病恶化逐步?炎症COPD可能逐步恶化,因为炎症和DNA损伤之间的恶性循环。3)为什么类固醇往往是无效的,除非加剧的情况下?类固醇对DNA损伤和上SASP [有限的效果没有影响的事实55]或许可以解释,至少部分,为什么类固醇往往是最COPD患者无效。4)为什么肺癌往往发展为COPD的并发症?开发用于肺癌的倾向作为COPD的并发症可能是由DNA损伤和氧化应激参与COPD和肺癌的发病机制中的事实来解释。5)为什么在COPD炎症甚至戒烟后坚持?炎症的慢性阻塞性肺病,甚至因为即使戒烟[后由吸烟引起的很长一段时间持续存在的DNA损伤戒烟后可能会持续56,57]。
结论
我们已经提出了一种两次打击假设暗示在COPD炎症变成慢性炎症的通过危险信号(第一命中)和炎症的DNA损伤的永久化(第二命中)的启动的结果。这一假说可以解释为什么慢性阻塞性肺病是中老年人(DNA损伤的积累)更常见。它也可以解释为什么炎症在COPD恶化逐步,为什么炎症在COPD甚至戒烟(DNA损伤的持久性)后仍然存在,以及为什么肺癌可以作为COPD的一种并发症(DNA损伤(炎症和DNA损伤之间恶性循环)和氧化应激促成COPD和肺癌)。
致谢
作者感谢s.i.r ennard(美国内布拉斯加大学医学中心,奥马哈,内布拉斯加大学医学中心,重症监护,睡眠和过敏科)对手稿的评论。
脚注
支持声明:该作品被支持的格兰特在急救从教育,科学,文化,日本的科研部和卫生部,劳动和日本福利的顽固性疾病的调查。
利益冲突:无申报。
- 收到了2012年7月4日。
- 公认二零一二年十二月十二日。
- ©ERS 2013











![Two-hit hypothesis, consisting of a danger signal plus DNA damage, explaining chronic inflammation in chronic obstructive pulmonary disease (COPD). a) In smokers, the danger signal (hit 1) is the trigger initiating inflammation in the lungs (the hypothesis of Cosio et al. [1]). According to the two-hit hypothesis, “susceptible” smokers who are predisposed to COPD develop DNA damage (hit 2), which induces apoptosis and senescence and activates the DNA damage response (DDR) and senescence-associated secretory phenotype (SASP), leading to the production of pro-inflammatory cytokines. b) Unlike healthy smokers, “susceptible” smokers develop DNA damage (e.g. double-strand breaks) in the airways and alveolar cells, presumably because of severer inflammatory cell infiltration and larger amounts of reactive oxygen species (ROS)/ reactive nitrogen species (RNS) production, a less efficient induction of antioxidants, and/or more inefficient DNA repair mechanisms. This, in turn, results in the activation of the DDR, apoptosis, senescence, and SASP and the production of pro-inflammatory cytokines. The pro-inflammatory cytokines then intensify inflammatory cell infiltration, forming a positive-feedback loop that further induces ROS/RNS production and the activation of the DDR, apoptosis, senescence, and SASP. In this manner, a vicious cycle is established between inflammation and DNA damage, and the inflammation becomes chronic. DAMP: damage-associated molecular pattern; IL: interleukin.](http://www.qdcxjkg.com/content/erj/42/6/1689/F1.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}