





摘要
肺动脉高压可发生在GATA2缺乏相关的肺泡蛋白沉积和纤维化http://ow.ly/aHv430aAZct
编辑器:
我们饶有兴趣地读了B的文章allerieet al。[1,描述了在GATA2缺乏症患者中肺泡蛋白沉积和纤维化的发生。有趣的是,我们的团队最近处理了一个类似的病例,其中间质性肺疾病最终导致肺动脉高压(PH)。
一名41岁女性于2014年因怀疑原发性免疫缺陷被转诊到我们的三级护理中心。既往病史包括复发性上呼吸道和下呼吸道感染、持续性血细胞减少(包括单核细胞减少、b细胞和自然杀伤细胞淋巴细胞减少)、弥散性分枝杆菌kansasii2002年感染和2008年骨髓增生异常综合征。她还表现出间质性肺病,2002年在胸部高分辨率计算机断层扫描(HRCT)中首次发现。2007年进行的肺活检显示肺泡蛋白沉积和肺旁纤维化的组织学病变。多年来,肺部疾病慢慢发展到累及整个肺实质。转诊时,胸部HRCT示双肺弥漫性小叶间隔增厚、网状、磨玻璃混浊(图1一个).总体情况与GATA2缺陷的诊断一致,该诊断于2014年10月被分子生物学证实。
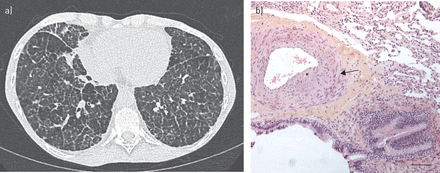
a)患者胸部高分辨率ct图像,双肺弥漫性小叶间隔增厚、网状、毛玻璃混浊。b)患者肺活检(苏木精和伊红染色)显示支气管血管束中肺动脉毗邻呼吸细支气管。动脉壁因中间平滑肌增生(箭头)和内膜纤维化(星号)而稍增厚,无血管炎。比例尺= 120μm。
在GATA2缺陷诊断时,患者抱怨她的呼吸困难缓慢恶化,并在纽约心脏协会功能分级IIa。体格检查未见右心衰迹象(包括下肢水肿)。6分钟步行试验证实,在不降低饱和度的情况下,步行距离为350米(预测值的53%),运动能力有限。胸部HRCT得到控制,显示稳定的间质异常,胸部螺旋ct排除肺栓塞。肺功能测试显示出限制性综合征(总肺活量为76%,强迫肺活量为预测值的55%),轻度低氧血症(室内空气中氧压81毫米汞柱,二氧化碳压29毫米汞柱)和一氧化碳肺扩散能力的显著变化(预测值的36%)。血清n端脑利钠肽前体水平在416 pg·mL轻微升高−1(正常情况下< 300 pg·毫升−1).经胸超声心动图显示PH(估计收缩期肺动脉压(PAP) 66mmhg)和右心室扩张(右-左心室直径比1.15),未见左心舒张期或收缩期功能不全。
2015年10月进行的右心导管检查显示中度毛细血管前PH值(收缩期/舒张期/平均PAP 48/17/32 mmHg,肺血管阻力6.0 Wood单位,肺动脉楔形压6mmhg,门静脉压力梯度正常),心功能保留(心输出量4.30 L·min−1心脏指数3.33 L·min−1·米−2).病人被认为患有酸碱度,原因是肺部疾病(世界卫生组织第三组[2])。因此,肺动脉高压的特异性治疗没有被引入。12个月后,临床、功能和超声指标保持稳定。
GATA2缺陷是一种以杂合突变为特征的新实体GATA2转录因子基因[3.].它与广泛的临床特征相关,包括非典型和/或复发性感染(特别是疱疹病毒、乳头瘤病毒和非结核分枝杆菌)、血细胞减少(特别是单核细胞减少)、血液系统恶性肿瘤(骨髓增生异常、急性骨髓母细胞白血病和慢性骨髓单核细胞白血病)、血管并发症(深静脉血栓形成和淋巴水肿)和间质性肺病(主要是肺泡蛋白沉积)[4].以往的刊物将酸碱度列为疾病可能的并发症之一[5- - - - - -7],发病率高达9% [6].然而,这些患者的详细特征(特别是在血流动力学方面)并没有被提供,而PH的确切机制也从未被研究。
在综合评估结束时,我们的患者被诊断为PH,由于长期的肺泡蛋白沉积和纤维化。由于GATA2在调节多种内皮细胞功能中发挥着关键作用[8,在这种情况下,另一个假定的PH机制也可能是特定的GATA2缺乏诱导的血管病变。在之前的工作中,Svobodovaet al。[9]描述了肺血管异常(动脉壁增厚,弹性层破坏和显示组织性脉管炎的血栓形成),该患者患有GATA2缺乏相关的肺间质性疾病。有趣的是,我们在2007年对患者(图1 b),超声心动图已显示与PH值相符的早期体征(估计收缩压PAP 43 mmHg)。这些异常是否与PH有关,无论是作为一个原因还是结果,仍然不确定。
GATA2缺乏症的治疗选择目前是有限的。最近的一项研究表明,异基因造血干细胞移植对6例患者的细胞减少和骨髓增生异常综合征可能有效[5].有趣的是,其中2例报告有PH值和肺泡蛋白沉积,移植后他们的呼吸状况得到改善[6].
综上所述,除了Ballerieet al。[1],医生应意识到GATA2缺乏症患者可能发生PH。
脚注
利益冲突:无声明。
- 收到了2017年1月24日。
- 接受2017年2月2日。
- 版权©2017人队











{kind=link}
{kind=link}