





摘要
特发性肺纤维化(IPF)是一种肺实质的进行性疾病,可导致严重的呼吸困难和整体功能下降。IPF的特征是抗凋亡的肌成纤维细胞,这是过度产生细胞外基质(ECM)超过正常肺组织的主要来源。我们试图研究热休克蛋白(HSP)异构体HSP90α和HSP90β的作用,它们在肺纤维化形成中的不同作用仍不清楚。
我们用ELISA法测定了31例IPF患者和9例年龄匹配的健康对照者的循环热休克蛋白90α水平。评估热休克蛋白90α和热休克蛋白90β的释放情况在体外在原发性IPF和对照肺成纤维细胞和体外对接受腺载体介导的转化生长因子-β1的大鼠肺纤维化切片进行机械拉伸。
我们证明,循环热休克蛋白90α在IPF患者中上调与疾病严重程度相关。纤维性ECM机械应力的增加促进了HSP90α的释放。这种细胞外HSP90α信号的增加通过低密度脂蛋白受体相关蛋白1 (LRP1)促进肌成纤维细胞的分化和持久性。同时,我们证明了细胞内形式的HSP90β稳定了LRP1,从而放大了HSP90α的细胞外作用。
我们认为,特异性抑制细胞外HSP90α是减少IPF中促纤维化信号的一种有前途的治疗策略。
摘要
细胞外HSP90α和细胞内HSP90β共同作用促进肺纤维化进展http://ow.ly/bAmH30gGQf1
简介
特发性肺纤维化(IPF)是一种肺实质的进行性疾病,可通过呼吸困难加重、咳嗽加重和整体功能下降引起显著发病率。IPF的确切病因仍然难以捉摸,治疗方案有限,只有两种获批准的IPF药物,即。Nintedanib和吡非尼酮被证明可以减缓但不能停止疾病进展[1,2].流行病学研究表明,在过去十年中,指规数的发病率一直在上升,特别是在>70岁年龄组[3.,4].IPF的预后较差,大多数患者死于疾病的比率与侵袭性癌症相当[5].IPF的特征是肺中抗凋亡肌成纤维细胞的增加,这是细胞外基质(ECM)异常沉积、转化生长因子(TGF)-β1等促纤维化细胞因子合成的主要来源,也与疾病进展有关[6].纤维性ECM比非纤维性ECM更硬,增加了纤维化肺内的机械应力[7].除了ECM的生化、结构和机械特征外,基质还是生长因子、细胞因子和各种影响周围细胞行为的细胞外蛋白的主要存储室。机械拉伸激活TGF-β1通过诱导肌成纤维细胞分化在IPF中发挥关键作用,从而增加纤维化肺中的ECM沉积[8].肌成纤维细胞分化是纤维发生的中心事件。在被称为上皮-间充质转化(EMT)的过程中,常驻成纤维细胞和上皮细胞被证明在TGF-β1刺激下分化为肌成纤维细胞[9].
HSP90属于应激诱导蛋白的热休克蛋白(HSP)家族。尽管HSPs具有细胞保护作用,但它们参与了癌症进展、转移形成和纤维化发展等病理过程[10].两种异构体,HSP90α和HSP90β,共享86%的氨基酸在所有有核细胞中表达。最近,HSP90α和较低程度的HSP90β已被证明由癌细胞和受各种应激条件(如缺氧、DNA损伤和生长因子刺激)的细胞分泌(如。转化生长因子-α)(11,12].细胞外HSP90已被报道能促进真皮成纤维细胞的细胞活力,加速小鼠皮肤伤口的愈合和再生上皮在活的有机体内[13].它也被证明是EMT的主要调节剂,促进前列腺癌模型中的转移形成[14].已有研究表明,低密度脂蛋白受体相关蛋白1 (LRP1)进一步激活细胞外信号调节激酶(ERK)和Akt磷酸化,并导致EMT,增加细胞活力和肌成纤维细胞分化,从而驱动细胞外HSP90活性[15,16].然而,HSP90α和HSP90β的细胞外形式的不同作用仍然不清楚。在这里,我们证明了循环中的HSP90α,而不是HSP90β,在IPF患者中与疾病严重程度相关的上调。纤维性ECM中机械应力的增加促进了HSP90α的释放。细胞外HSP90α的增加通过受体LRP1激活肺上皮细胞和成纤维细胞中的促纤维化途径,并促进基质重构,从而建立促纤维化微环境,允许肌成纤维细胞分化和持久性。同时,细胞内形式的HSP90β稳定LRP1,从而放大HSP90α的细胞外作用。因此,我们认为特异性抑制细胞外HSP90α是减少IPF中促纤维化信号的一种有前途的治疗策略。
材料与方法
看到补充材料详细方法。
人类的样本
在汉密尔顿综合研究伦理委员会(00-1839)的患者批准下收集所有血浆和组织。对照组的肺组织来自接受癌症手术的患者。从接受活检的患者中收集肺纤维化组织,以诊断不明确的间质性肺疾病。本研究的活检结果显示组织病理学上为间质性肺炎(UIP)。
动物的过程
Sprague Dawley大鼠(8周龄)按照加拿大动物护理委员会的指南进行治疗,并经麦克马斯特大学动物研究伦理委员会批准(议定书13.12.48)。大鼠收到了5.0×108如前所述,在异氟醚麻醉下气管内灌注腺载体介导的TGF-β1 (AdTGF-β1)(或对照AdDL)的PFU补充的方法有关详情)[17].
细胞培养
对照和IPF原代肺成纤维细胞如前所述从3例接受癌症手术的患者的无瘤肺区和3例接受手术并最终确认为IPF- uip的间质性肺疾病患者中分离出来[19].原代肺成纤维细胞在DMEM/10%胎牛血清(FBS)中培养。A549细胞(ATCC, Teddington, UK)在DMEM/10%胎牛血清中培养。细胞在常规组织培养板(Thermo Fisher Scientific, Burlington, on, Canada)或涂有不同硬度水凝胶的组织培养板(Matrigen, Brea, CA, USA;1kpa(软)或50kpa(硬))。当提示时,人原代肺成纤维细胞(对照组和IPF)在常规组织培养板上培养(以允许膨胀)直到第2代,然后在硬或软的水凝胶上培养到第3代,用于实验。当有提示时,用HS-30的5µM(细胞处理)或10µM(共免疫沉淀)处理细胞,HS-30是一种专门设计为细胞不渗透的HSP90抑制剂。HS-30是一种HSP90抑制剂衍生物,设计如上所述,由Timothy Haystead(杜克大学药理学和癌症生物学系,美国北卡罗来纳州达勒姆)提供[20.,21].MG132 (Sigma Aldrich, Oakville, ON, Canada)抑制蛋白酶体活性,并在细胞收获前6小时以50 μ M的浓度使用。利用小干扰RNA (HSP90α: s6994;一半β:s14375;LRP1恰巧:s8279;赛默飞世尔科学公司)。重组(r) HSP90α (Enzo Life Science, Farmingdale, NY, USA)以10 μ M的浓度处理A549细胞和原代成纤维细胞24-48 h。rTGF-β1 (R&D Systems, Minneapolis, MN, USA)以2 ng·mL的浓度处理A549细胞48小时−1.用阻断型HSP90α抗体(AbHSP90α)以40 μ M的浓度处理A549细胞和原代成纤维细胞24-48 h。用TGF-β受体I (TGF-β ri)抑制剂SD-208在30 μ M浓度下处理原代成纤维细胞24-48 h。
西方墨点法
Western blotting法检测热休克蛋白90α (ADI-SPS-771-F;Enzo Life Science), HSP90β (ADI-SPA-844-200;TGF-β1 (ab92486;Abcam, Toronto, ON, Canada), α-平滑肌肌动蛋白(α-SMA) (ab7817;增殖细胞核抗原(ab18197;Abcam),磷酸化Smad2 (p-Smad2) (#3101;细胞信号技术,Beverly, MA, USA), Smad2 (ab63576;Abcam),胶原蛋白1A1 (ab34710;Abcam), ERK (#9102;细胞信号技术),磷酸化ERK (p-ERK) (#4377; Cell Signaling Technology), LRP1 (ab92544; Abcam), Na,K-ATPase (#3010; Cell Signaling Technology), TGF-βRI (ab31013; Abcam) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (ab9485; Abcam). See补充的方法获取详细信息。
免疫荧光
热休克蛋白90α (ab59459;Abcam)和LRP1 (ab92544;Abcam)在福尔马林固定的人体肺组织切片上进行(见补充的方法详情)。
体外机械拉伸程序
大鼠纤维肺切片采用我们的机械拉伸方案,如前所述(补充数字S1) [22].在条状组织被拉伸之前,从组织浴中提取的样本被称为“之前”。为了得到杨氏模量的测量值,在10到15秒之间应用了静态5 mN。在这些测量之后,进行4分钟的15 mN循环拉伸,并从组织浴溶液中收集样本(称为“之后”)。未拉伸的肺组织被称为“未受刺激”,拉伸的肺组织被称为“受刺激”。
我们的目的是通过设置循环拉伸张力为15 mN,以2 Hz的频率振荡4分钟来模拟组织浴中的呼吸张力,长度为组织条(测量到的肺条10×2×2 mm)原始静止长度的1.1倍。估计压力为每4毫米15 mN2= 0.37 mN·毫米2= 37.73而言不啻2O,在IPF患者肺部观察到的压力范围内[23].在一些实验中,用蛋白质转运抑制剂预处理肺切片2小时(eBioscience, San Diego, CA, USA)。组织刚度用杨氏模量(N·mm)表示2)如上文所述[22].看到补充的方法获取详细信息。
体外抑制细胞外HSP90
在第21天收集接受AdTGF-β1的大鼠肺纤维化切片。肺切片在DMEM/10%胎牛血清中培养,接受HS-30 (10 μ M)或载药培养72 h。采集肺切片进行mRNA和蛋白表达分析。
免疫共沉淀(膜分离)
对于LRP1共免疫沉淀,使用试剂盒(ab65400;(Abcam)按照制造商的建议。采用ExactaCruz试剂盒(Santa Cruz Biotechnology, Dallas, TX, USA)按照制造商的建议对质膜组分或全细胞提取物进行免疫共沉淀实验。看到补充的方法获取详细信息。
统计分析
所有数据均以平均值±表示扫描电镜.两组间的统计分析采用参数法t以及。多组间统计分析,其中一个对照组采用单因素方差分析,采用Tukey多重比较检验(事后).采用Prism 6.0版本(GraphPad, La Jolla, CA, USA)进行分析。p值<0.05被认为是显著的。
结果
循环中的HSP90α在IPF患者和AdTGF-β1诱导的肺纤维化中上调
测定IPF患者(n=31)和年龄匹配对照组(n=9)血清中循环HSP90α和HSP90β水平。与对照组相比,IPF患者中HSP90α显著上调,而HSP90β保持不变(图1一个而且补充图S2).为了进一步表征热休克蛋白90α在IPF中的上调,根据患者的用力肺活量(FVC) % pred将患者分为两组:根据文献将中度IPF (FVC % pred >60%, n=25)和重度IPF (FVC % pred <60%, n=6) [24].有趣的是,与对照组相比,中重度IPF患者循环中HSP90α表达上调;重度IPF患者循环热休克蛋白90α水平明显高于中度IPF患者(图1一个).HSP90β在两组间无差异(补充图S2).此外,IPF患者血清热休克蛋白90α水平与肺功能测试:总肺活量、FVC和1 s用力呼气量(图1 b).
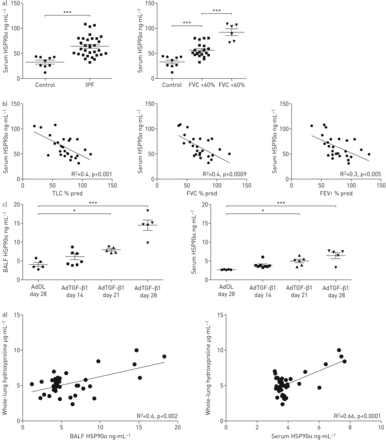
循环热休克蛋白(HSP)亚型HSP90α在特发性肺纤维化(IPF)患者和腺载体介导的转化生长因子-β1 (AdTGF-β1)诱导的肺纤维化中表达上调。a)用ELISA法测定IPF患者和相应年龄匹配的健康志愿者血清热休克蛋白90α水平。数据以平均值±表示扫描电镜;对照组n=9, IPF组n=31。IPF患者分为两组:中度IPF (FVC % pred >60%)和重度IPF (FVC % pred <60%)。数据以平均值±表示扫描电镜;对照组、中度IPF和重度IPF患者n=9、n=25和n=6。b) IPF患者血清HSP90α水平与临床肺功能参数:总肺活量(TLC)、用力肺活量(FVC)和1 s用力呼气量(FEV)的相关性曲线1).c) AdTGF-β1(或AdDL作为对照)处理大鼠的支气管肺泡灌洗液(BALF)和血清HSP90α水平。数据以平均值±表示扫描电镜;AdDL: n = 5;AdTGF-β1 d 14: n=8;AdTGF-β1 day 21: n=5;AdTGF-β1天28:n=5。d) AdTGF-β1处理大鼠BALF与血清HSP90α水平和羟脯氨酸肺含量的相关性曲线。*: p < 0.05;* * *: p < 0.001。
大鼠气管内接受AdTGF-β1后,在注射后第28天发生强烈的进行性纤维化[17].Western blot分析显示,经AdTGF-β1治疗的纤维化大鼠支气管肺泡灌洗液(BALF)中HSP90α水平在第28天上调。补充图S2).这一结果在BALF和AdTGF-β1处理大鼠血清的ELISA检测中得到证实。图1 c).有趣的是,从第14天(早期纤维化)到第28天(晚期纤维化),细胞外HSP90α水平逐渐升高,表明HSP90α水平与纤维化进展之间存在相关性。事实上,在AdTGF-β1处理的大鼠BALF和血清中HSP90α的细胞外水平与羟脯氨酸的数量相关,羟脯氨酸是纤维化过程中胶原蛋白增加的间接量化指标,肺纤维化评分(Ashcroft评分)(图1 d而且补充图S2).
总之,这些结果表明,循环中的HSP90α,而不是HSP90β,在IPF患者和肺纤维化大鼠中上调与纤维化严重程度相关。
机械拉伸和组织僵硬诱导HSP90α分泌
首先评估HSP90α和HSP90β的分泌在体外来自对照组和IPF患者的肺上皮细胞、A549细胞和原代肺成纤维细胞。与对照组相比,IPF成纤维细胞在没有任何刺激的情况下分泌HSP90α上调,而A549细胞即使在TGF-β1刺激后也没有分泌任何HSP90α (图2一个).对原代成纤维细胞和A549细胞上清进行Western blot分析,证实了上述结果。补充图S3).A549细胞或原代成纤维细胞未见HSP90β分泌(图2一个而且补充图S3).与正常肺组织(范围1kpa)相比,常规组织培养板非常坚硬(范围1gpa)。因此,我们将原代成纤维细胞培养在不同硬度的水凝胶上,以测量HSP90α的分泌。软性水凝胶的硬度为1千帕(相当于正常肺),硬性水凝胶的硬度为50千帕(相当于纤维化肺)[7].在坚硬的水凝胶和常规的组织培养板上,IPF成纤维细胞中HSP90α的分泌量高于对照组。然而,IPF成纤维细胞分泌HSP90α在软底物上受到抑制(图2 b).原代成纤维细胞在软或硬的水凝胶上不分泌HSP90β (图2 b).在IPF中与疾病进展相关的另一个重要刺激是机械拉伸。随着疾病的进展,ECM沉积的增加导致肺组织硬度的增加,这反过来加剧了呼吸时的机械拉伸,有利于IPF进展[22].我们的团队开发了一个模型,模拟IPF患者呼吸时发生的机械拉伸(补充图S1).该模型允许应用机械拉伸对纤维化肺切片和测量体外机械拉伸刺激后组织硬度和分泌蛋白的定量[22].利用该模型,我们证明了机械拉伸诱导AdTGF-β1处理大鼠的纤维化肺切片释放HSP90α,但不释放HSP90β,而非纤维化肺切片不释放HSP90α或HSP90β (图2 c).此外,热休克蛋白90α的释放与纤维化肺切片的刚度(杨氏模量)呈正相关(补充图S3).有趣的是,用细胞分泌抑制剂预处理纤维化肺切片完全消除了机械拉伸引起的HSP90α的释放(图2 d).
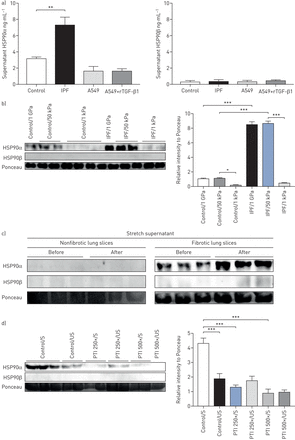
机械拉伸和组织僵硬诱导热休克蛋白(HSP)异构体HSP90α的分泌。a)重组转化生长因子(rTGF)-β1处理或未处理的对照组/原发性特发性肺纤维化(IPF)肺成纤维细胞和A549细胞上清中HSP90α和HSP90β的水平。将细胞置于无血清培养基24 h后,用ELISA法测定热休克蛋白90α和热休克蛋白90β。数据以平均值±表示扫描电镜;n = 3。b) Western blot检测上清中HSP90α和HSP90β的表达(相应的HSP90α密度分析)。细胞分别在硬度为1kpa(软)或50kpa(硬)的水凝胶或常规组织培养板(1gpa)上培养,并置于无血清培养基中24 h后收集上清。Ponceau染色作为加载对照。数据以平均值±表示扫描电镜;n = 4。c) Western blot分析拉伸前后非纤维化和纤维化肺切片(来自腺载体介导的转化生长因子-β1 (AdTGF-β1)治疗大鼠)中HSP90α和HSP90β表达的释放情况。Western blots是三个独立实验的代表。在分离的凝胶上对纤维化和非纤维化肺切片进行Western blot,但同时进行。Ponceau染色作为加载对照。d) Western blot分析纤维肺切片(来自AdTGF-β1治疗的大鼠)在拉伸前后加或不加蛋白转运抑制剂(PTI)治疗后HSP90α和HSP90β表达的释放情况(并对HSP90α进行相应的密度分析)。S:刺激;我们:如果。Ponceau染色作为加载对照。 Data presented as mean±扫描电镜;n = 6。*: p < 0.05;* *: p < 0.01;* * *: p < 0.001。
总之,这些结果表明,在组织硬化和机械拉伸刺激后,IPF肺成纤维细胞分泌HSP90α,而不是HSP90β。
细胞外HSP90α诱导肌成纤维细胞分化
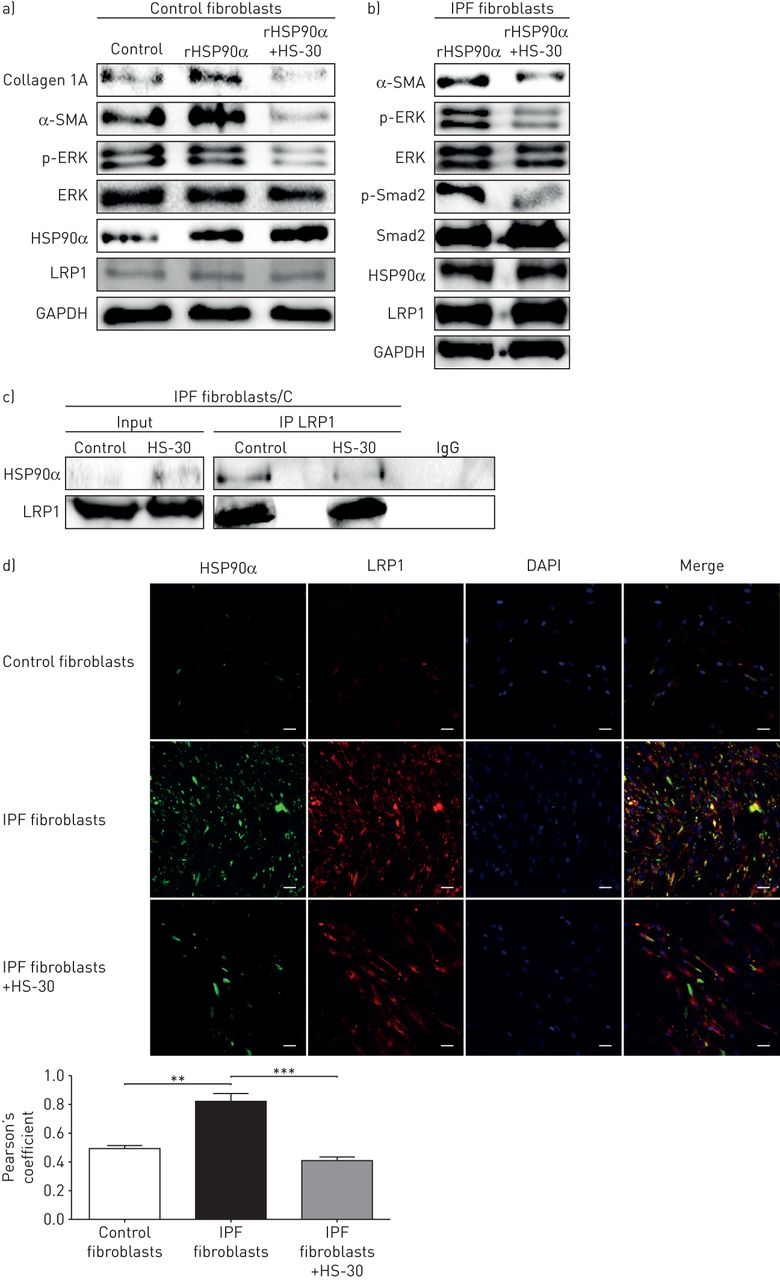
用rHSP90α治疗肺上皮细胞和对照成纤维细胞诱导了主要促纤维化标志物的上调。事实上,rHSP90α促进了肌成纤维细胞标记物α-SMA的表达,并诱导了TGF-β1通路通过Smad2磷酸化以及ERK通路。反过来,rHSP90α促进了胶原蛋白1A (图3一有趣的是,在A549细胞中,rHSP90α上调α-SMA和下调上皮标记物E-cadherin提示EMT的诱导(图3一).用rHSP90α治疗IPF成纤维细胞并没有引起促纤维化标记物的表达变化,而这些标记物已经高度表达(补充图S4).相比之下,阻断抗体抑制HSP90α可降低IPF成纤维细胞中α-SMA和胶原1A的表达以及ERK磷酸化(图3 c).有趣的是,来自IPF成纤维细胞的条件培养基有利于上皮细胞的迁移,而不是来自对照成纤维细胞的条件培养基。这种迁移被阻断抗体抑制HSP90α (图3 d).
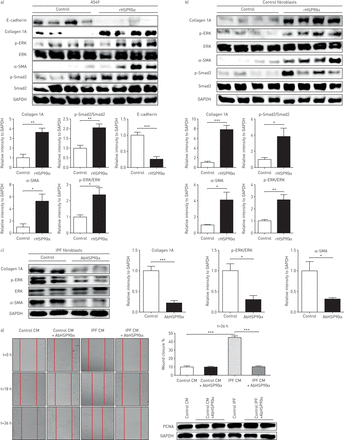
细胞外热休克蛋白(HSP)异构体HSP90α诱导肌成纤维细胞分化。a)在重组(r) HSP90α或载药(对照)处理的A549细胞上,对e -钙粘蛋白、1A胶原蛋白、磷酸化的细胞外信号调节激酶(p-ERK)、ERK、α-平滑肌肌动蛋白(α-SMA)、磷酸化的Smad2 (p-Smad2)和Smad2进行Western blot分析(及相应的密度测定法)。甘油醛3-磷酸脱氢酶(GAPDH)染色作为加载对照。数据以平均值±表示扫描电镜;n = 4。b) rHSP90α或对照剂(对照组)处理人原代对照成纤维细胞48h后,Western blot分析1A、p-ERK、ERK、α-SMA、p-Smad2和Smad2胶原蛋白(及相应的密度测定)。GAPDH染色作为加载对照。数据以平均值±表示扫描电镜;n = 4。c)阻断型HSP90α抗体(AbHSP90α)或对照剂(对照组)处理的人原发性特发性肺纤维化(IPF)成纤维细胞的1A胶原蛋白、p-ERK、ERK和α-SMA蛋白的Western blot分析(及相应的密度测定)。GAPDH染色作为加载对照。数据以平均值±表示扫描电镜;n = 4。d) A549细胞用对照或IPF原代成纤维细胞的条件培养基(CM)培养,用AbHSP90α或载物(对照组)处理,在40 μ M。图像是三个独立实验的代表。该图表示伤口治疗后36小时测量的伤口闭合百分比。A549细胞对应的增殖细胞核抗原(PCNA)表达在伤口闭合试验中未见增殖增加。数据以平均值±表示扫描电镜;n = 3。*: p < 0.05;* *: p < 0.01;* * *: p < 0.001。
综上所述,这些结果表明HSP90α促进肺上皮细胞的EMT和成纤维细胞向肌成纤维细胞的分化,而其抑制作用限制了IPF成纤维细胞的侵袭表型。
HSP90α信号通过受体LRP1
已有研究表明,细胞外HSP90活性是由受体LRP1驱动的[15].令人惊讶的是,我们无法从对照或IPF成纤维细胞的质膜蛋白提取物中共同免疫沉淀HSP90α和LRP1 (图4一).但HSP90β和LRP1共免疫沉淀较强(图4一).我们假设HSP90α和LRP1之间的细胞外结合可能太弱,不足以显示阳性的共免疫沉淀。因此,在进行免疫共沉淀之前,我们用蛋白质交联剂对细胞进行预处理。在对照组成纤维细胞中,HSP90α和LRP1在没有交联或有交联的情况下没有共同免疫沉淀(图4 b),而HSP90α和LRP1在交联后IPF成纤维细胞中共免疫沉淀(图4 b).因此,我们假设HSP90α和LRP1之间的结合只存在于细胞外(只在交联后可见),而HSP90β和LRP1之间也有意想不到的结合存在于细胞内(强免疫共沉淀而不交联)。由于HSP90β在细胞内的作用主要是蛋白质伴侣(防止降解),我们使用特定的siRNA抑制IPF成纤维细胞内的HSP90β。抑制HSP90β可引起LRP1表达的强烈下调。当用蛋白酶体降解途径抑制剂MG132处理细胞时,LRP1的表达恢复。图4 c).然而,细胞内抑制HSP90α并没有引起LRP1的下调(图4 c).
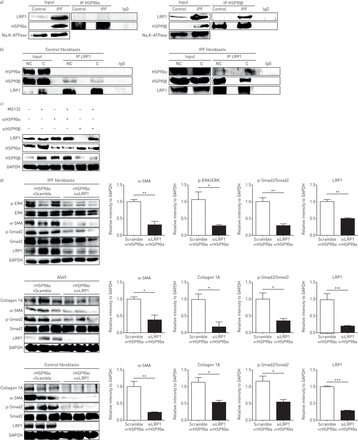
热休克蛋白(HSP)亚型HSP90α信号通过低密度脂蛋白受体相关蛋白1 (LRP1)传递。a)对质膜提取的蛋白进行HSP90α和HSP90β的免疫沉淀(IP),然后对LRP1和HSP90α进行免疫检测。对照:人原代成纤维细胞对照;IPF:特发性肺纤维化人原代成纤维细胞;IgG:非相关抗体;输入:非免疫沉淀提取物。Na、k - atp酶作为质膜组分的负载控制。b)在质膜提取的蛋白上免疫沉淀LRP1,然后免疫检测HSP90α、HSP90β和LRP1。免疫沉淀与细胞外/膜蛋白预先交联或不交联进行:对照人原代成纤维细胞和人原代IPF成纤维细胞。NC: non-cross-linked; C: cross-linked. c) Western blot analysis of LRP1, HSP90α and HSP90β expression on IPF human primary fibroblasts treated with small interfering (si) RNA for HSP90α or HSP90β (Scramble siRNA used as control) and treated with or without MG132 for 6 h at 50 µM. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) staining served as loading control. The Western blot is representative of three independent experiments. d) Western blot analysis of phosphorylated extracellular signal-regulated kinase (p-ERK), ERK, α-smooth muscle actin (α-SMA), phosphorylated Smad2 (p-Smad2), Smad2, LRP1 and collagen 1A expression on human primary IPF fibroblasts, A549 cells and control fibroblasts treated with siRNA for LRP1 (Scramble siRNA used as control) and treated with recombinant (r) HSP90α for 48 h at 10 µM. GAPDH staining served as loading control. Data presented as mean±扫描电镜;n = 3。*: p < 0.05;* *: p < 0.01;* * *: p < 0.001。
为了确认涉及HSP90α和LRP1的信号通路,我们用rHSP90α处理肺上皮细胞、对照细胞和IPF成纤维细胞,而抑制LRP1通过一个核。有趣的是,LRP1的抑制通过阻止α-SMA和胶原蛋白的上调以及Smad2和ERK的磷酸化,在所有类型的细胞中完全废除了rHSP90α的作用(图4 d).
综上所述,这些结果表明HSP90α信号通过LRP1,细胞内增强了LRP1的稳定性。
HS-30是细胞外HSP90的抑制剂,可抑制HSP90α/LRP1信号通路
HS-30是HSP90的选择性抑制剂,经修饰后具有细胞渗透性[20.].HS-30处理对照组和IPF成纤维细胞抑制rHSP90α对这些细胞的影响,通过抑制α-SMA和胶原蛋白的上调以及Smad2和ERK的磷酸化(图5一个有趣的是,HS-30抑制交联IPF成纤维细胞中HSP90α和LRP1的共免疫沉淀(图5 c).免疫荧光证实了这一结果。与对照组相比,IPF成纤维细胞中HSP90α和LRP1的共域增强。HS-30显著抑制HSP90α和LRP1的共化(图5 d).为了进一步证实人类HSP90α和LRP1之间的联系,我们对人类肺组织进行了免疫荧光。中重度IPF患者与对照组相比,HSP90α和LRP1共灶明显升高(图6).
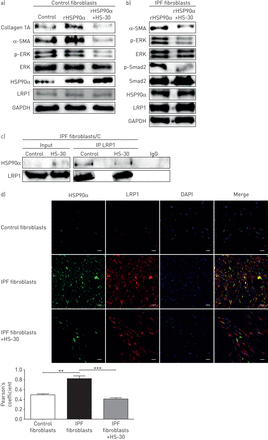
HS-30是细胞外热休克蛋白HSP90的抑制剂,可阻止HSP90α/低密度脂蛋白受体相关蛋白1 (LRP1)的信号传导。a) Western blot分析重组(r) HSP90α处理的人原代对照成纤维细胞上胶原1A、α-平滑肌肌动蛋白(α-SMA)、磷酸化细胞外信号调节激酶(p-ERK)、ERK、HSP90α和LRP1的表达,10 μ M,含或不含HS-30, 1 mM。甘油醛3-磷酸脱氢酶(GAPDH)染色作为加载对照。Western blot是三个独立实验的代表。b) Western blot分析人原发性特发性肺纤维化(IPF)成纤维细胞中α-SMA、p-ERK、ERK、磷酸化Smad2 (p-Smad2)、Smad2、HSP90α和LRP1的表达,以10 μ M,含或不含HS-30, 1 mM为µM。GAPDH染色作为加载对照。Western blot是三个独立实验的代表。c)在质膜提取的蛋白上进行LRP1的免疫沉淀(IP),然后进行HSP90α和LRP1的免疫检测。C:交联;输入:非免疫沉淀提取物; IgG: nonrelevant antibody. Immunoprecipitation was performed with prior cross-linking of extracellular/membrane proteins. Immunoprecipitation performed on IPF fibroblasts treated with or without HS-30 at 1 mM. d) Immunofluorescence of LRP1 (red) and HSP90α (green) on control fibroblasts, IPF fibroblasts and IPF fibroblasts treated with HS-30 at 1 mM. Nuclear staining: 4′,6-diamidino-2-phenylindole (blue). Images are representative of three independent experiments. Scale bar: 50 μm. Pearson's coefficient for each condition presented as mean±扫描电镜;n = 3。* *: p < 0.01;* * *: p < 0.001。
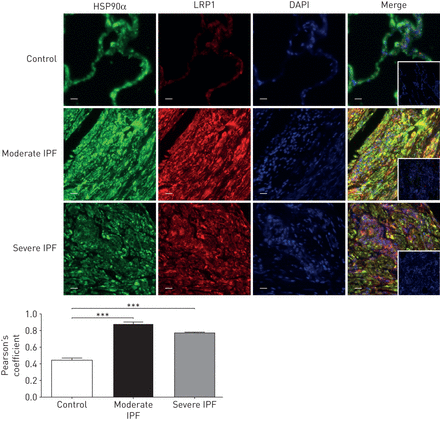
热休克蛋白(HSP)亚型HSP90α/低密度脂蛋白受体相关蛋白1 (LRP1)在特发性肺纤维化(IPF)患者中共聚集。LRP1(红色)和HSP90α(绿色)在对照组、中度IPF患者和重度IPF患者肺组织中的免疫荧光。IgG作为插入物。核染色:4 ',6-二氨基-2-苯基吲哚(蓝色)。所展示的图像是三个独立实验的代表。比例尺:50 μm。每种条件的皮尔逊系数用平均值±表示扫描电镜;n = 3。* * *: p < 0.001。
IPF成纤维细胞中的HSP90α信号与TGF-β1通路无关
细胞外热休克蛋白90α最近被证实与TGF-βRI结合,促进TGF-β激活的心脏成纤维细胞胶原蛋白的生成[25].在我们的模型中,IPF成纤维细胞中的细胞外HSP90α即使在交联(补充图S5).此外,TGF-βRI抑制剂SD-208对肺成纤维细胞中α-SMA和TGF-β1的增加以及rHSP90α诱导的ERK磷酸化没有抑制作用(补充图S6).同样,rHSP90α诱导TGF-β1表达和激活增加,SD-208不抑制,而HS-30抑制。补充图S6).
综上所述,这些结果表明HSP90α信号通路独立于TGF-β1通路。
HS-30对纤维化肺组织具有抗纤维化特性体外
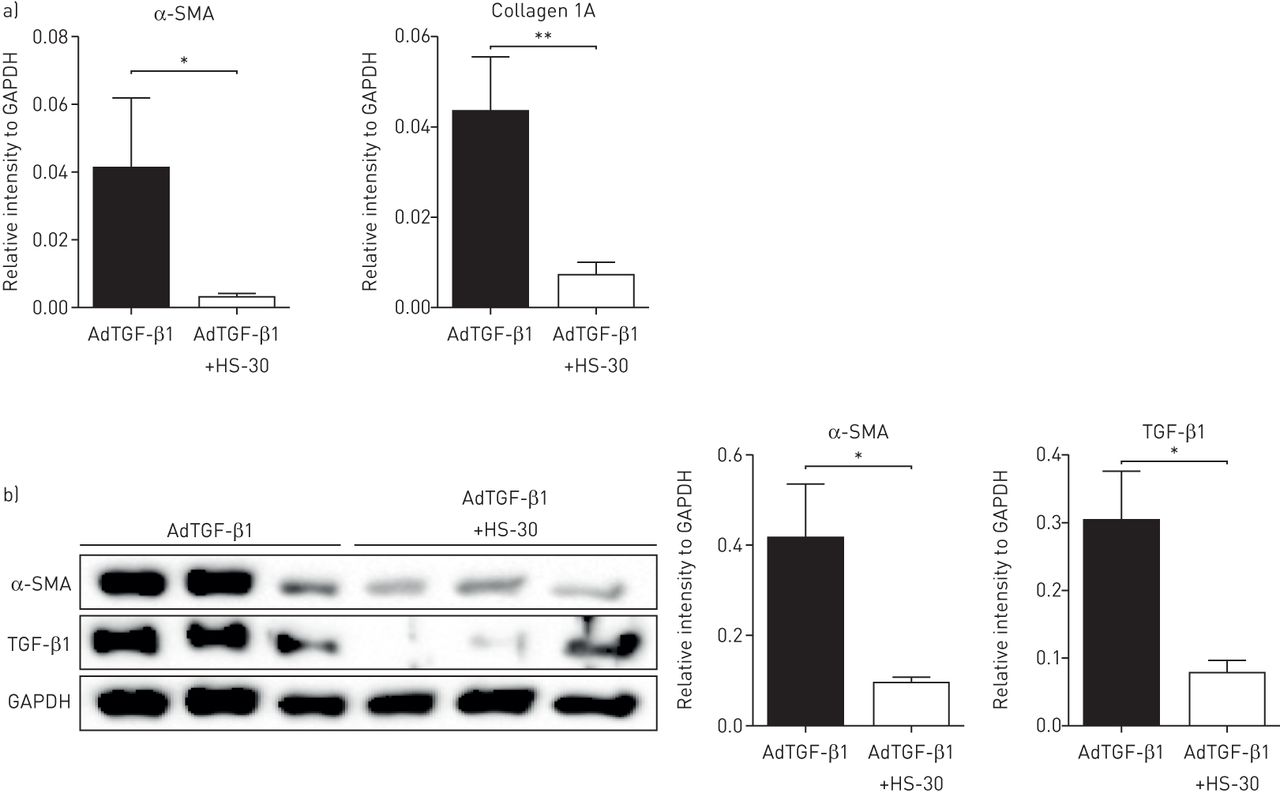
第21天AdTGF-β1处理动物的纤维肺切片用HS-30或对照剂处理72 h体外.与对照组相比,HS-30可诱导AdTGF-β1处理动物肺切片中胶原蛋白和α-SMA mRNA表达的降低(图7).此外,HS-30还降低了α-SMA和TGF-β1蛋白的表达,显示了细胞外HSP90对纤维化肺组织肌成纤维细胞分化和持久性的抑制作用(图7 b).
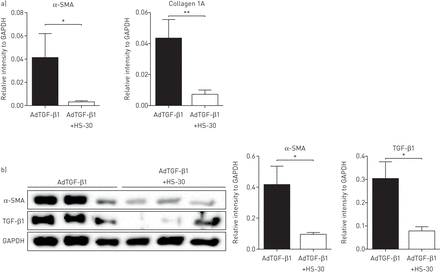
HS-30对纤维化肺组织具有抗纤维化特性体外.a)接受腺载体介导的转化生长因子-β1 (AdTGF-β1)的大鼠肺切片上α-平滑肌肌动蛋白(α-SMA)和胶原1A表达的定量反转录PCR分析。AdTGF-β1给药后第21天收集肺切片,进行培养体外使用HS-30或车辆72小时。数据以平均值±表示扫描电镜;n = 4。b)接受AdTGF-β1的大鼠肺切片上α-SMA和TGF-β1表达的Western blot分析(及相应的密度测定法)。AdTGF-β1给药后第21天收集肺切片,进行培养体外使用HS-30或车辆72小时。甘油醛3-磷酸脱氢酶(GAPDH)染色作为加载对照。数据以平均值±表示扫描电镜;n = 4。*: p < 0.05;* *: p < 0.01。
讨论
IPF的特征是出现抗凋亡的肌成纤维细胞,过度产生异常的ECM,导致肺组织硬化和疾病进展。因此,IPF被视为一种不受控制的伤口愈合反应,其中许多因素阻碍了形成适当瘢痕组织所需的肌成纤维细胞的清除。在IPF的情况下,肌成纤维细胞的持续存在使疤痕组织堆积,逐渐破坏正常的肺结构,损害肺功能[26].TGF-β1在肌成纤维细胞分化中的中心作用已被多次证实[6,8].在目前的研究中,我们发现了另一种循环蛋白HSP90α,它能够诱导肺上皮细胞和肺成纤维细胞向肌成纤维细胞分化。有趣的是,在肺纤维化动物模型中,循环热休克蛋白90α水平与胶原沉积相关,也与IPF患者的疾病严重程度相关。需要对更多IPF患者和其他非IPF间质性肺疾病患者进行进一步研究,以评估循环热休克蛋白90α作为患者IPF严重程度和IPF进展的生物标志物的潜力。
我们的结果强调,促纤维化HSP90α的分泌是由组织硬度和机械拉伸驱动的。由于瘢痕组织的积累,IPF肺比正常肺更硬[7].因此,IPF肺在呼吸时承受较高的机械拉伸[23].IPF肺中硬度和机械拉伸力的增加已经被证明与纤维化进展有关,主要是通过TGF-β1的激活,而TGF-β1反过来激活肌成纤维细胞,产生更多异常的ECM [22].在我们的研究中,我们证明了,伴随着TGF-β1的激活,机械拉伸作用于纤维化肺切片诱导HSP90α释放到细胞外空间,这也能够激活肌成纤维细胞分化。我们进一步证明了HSP90α信号通过受体LRP1促进肌成纤维细胞分化。令人惊讶的是,虽然HSP90β没有分泌,但它在细胞内与LRP1结合,从而稳定受体并进一步激活HSP90α/LRP1信号。这些发现与在伤口愈合模型中得到的结果一致,在该模型中,细胞外HSP90已被证明通过LRP1发出信号,促进皮肤细胞迁移在体外而且在活的有机体内[27].此外,Jayaprakash等.[28研究表明,热休克蛋白90α和热休克蛋白90β共同促进受伤皮肤的细胞运动,从而加速伤口愈合。在他们的模型中,HSP90β稳定了LRP1,而分泌的HSP90α通过LRP1发出信号[28].最近,Sontake等.[29研究表明,HSP90α和HSP90β在IPF中具有不同的作用。在他们的研究中,HSP90β的细胞内特异性抑制减弱了原纤维基因的表达,如胶原蛋白1α,胶原蛋白5α和α-SMA基因。有趣的是,作者证明了细胞内抑制HSP90α对促纤维化信号通路没有影响。这些结果与我们的研究一致,我们清楚地证明了HSP90β的促纤维化作用是在细胞内介导的,而HSP90α的促纤维化作用是在细胞外介导的。在IPF中,肺组织硬度的增加可能通过刺激HSP90α的分泌过度激活该通路,导致伤口愈合失控和进行性纤维化。这些结果表明,HSP90α的过度分泌和肺硬度的增加都是HSP90α作为促纤维化信号持续活跃的来源。因此,同时抑制细胞外热休克蛋白90α,减轻肺硬化是一种有效的治疗策略。这种策略在硬皮病相关肺部疾病的动物模型中显示出有希望的结果。ECM交联可放大IPF肺组织的硬度[30.].l在等.[31]证明了一种使用松弛素对抗肌成纤维细胞收缩能力的治疗只有在与靶向基质交联的治疗结合以限制肺僵硬时才有效。可以想象,抑制HSP90α细胞外信号,结合同种旨在减少肺僵硬的疗法,可能是一种很有前景的方法,以对抗已经建立的纤维化。
有趣的是,我们在本研究中描述的HSP90α细胞外信号似乎独立于主要的促纤维化途径TGF-β1。细胞内HSP90已被证实可稳定TGF-βRII,促进肺和肾纤维化中的肌成纤维细胞分化[32- - - - - -35].此外,细胞外HSP90最近被证实与TGF-βRI结合,并参与TGF-β1诱导的心肌成纤维细胞胶原生成[25].然而,在我们的人原代肺成纤维细胞模型中,细胞外HSP90α的作用不能被SD-208抑制,SD-208是TGF-βRI的特异性抑制剂。由于IPF成纤维细胞表达高水平的LRP1,我们认为HSP90α/LRP1信号至少部分驱动了肌成纤维细胞的持久性。最近的研究结果表明,在IPF患者中HSP90表达上调,治疗性给药HSP90抑制剂17-N-烯丙胺-17-去甲氧基格尔丹霉素(17-AAG)可抑制博莱霉素诱导的肺纤维化,突出了在IPF中抑制HSP90的潜在益处[29].然而,17-AAG靶向的是HSP90α和HSP90β细胞内活性。根据本文的结果,我们认为IPF中针对细胞外HSP90α信号的特异性靶向可能有助于克服完全抑制细胞内和细胞外HSP90的细胞保护功能可能导致的问题。没有具体的在活的有机体内细胞外热休克蛋白90α抑制剂的存在,这在一定程度上限制了我们的研究在活的有机体内肺纤维化过程中细胞外热休克蛋白90抑制的作用。然而,我们的研究结果表明,抑制细胞外HSP90通过减少胶原蛋白表达和肌成纤维细胞分化和持久性显示出有前景的抗纤维化作用体外.因此,这项新的实验研究以及来自实验和临床研究的越来越多的证据使热休克蛋白,特别是热休克蛋白90α和热休克蛋白90β越来越成为IPF药物开发的重点。
补充材料
补充材料
请注意:补充材料不由编辑部编辑,上传时由作者提供。
补充材料erj - 00386 - 2017 - _supplement
图S1。机械拉伸机。a)自顶向下的示意图表示体外模型应用机械拉伸肺条。b)照片体外模型应用机械拉伸。erj - 00386 - 2017 - _figure_s1
图S2。循环中的HSP90α在IPF患者和AdTGF-β1诱导的肺纤维化中上调。a)用ELISA法测定IPF患者和相应年龄匹配的健康志愿者的血清HSP90β水平(左图)。结果以平均值±SEM表示,对照组和IPF患者分别为n=9和n=31。IPF患者被分为两组(右图):中度IPF (FVC %预测>为60%)和重度IPF (FVC %预测<60%)。结果以平均值±SEM表示。***p<0.001, n=9, n=25, n=6分别为对照组、中度IPF和重度IPF患者。b) Western blot分析AdTGF-β1处理大鼠BALF中HSP90的表达(AdDL=对照组)。提出的WB具有3个独立实验的代表性。c) AdTGF-β1处理大鼠血清中HSP90α与BALF水平与相应肺Ashcroft评分的相关性曲线。erj - 00386 - 2017 - _figure_s2
图S3。机械拉伸和组织僵硬诱导HSP90α分泌。A. Western blot分析未处理对照/IPF原代肺成纤维细胞和未用rTGF-β1处理的A549细胞上清中HSP90α和HSP90β的表达。给出的WB是3个独立实验的代表。Ponceau染色作为加载对照。b)纤维肺条拉伸后释放的热休克蛋白90α水平与相应肺条Youngâs模量(刚度)的相关曲线。erj - 00386 - 2017 - _figure_s3
图S4。在经rHSP90α或对照剂(ctrl)在10 μM条件下处理48小时的人原代IPF成纤维细胞中,Western blot分析(和相应的密度测定)1A、p-ERK、ERK、p-Smad2、Smad2和α-SMA胶原蛋白。GAPDH染色作为加载对照。结果以平均值±SEM表示,n=4。erj - 00386 - 2017 - _figure_s4
图S5。rHSP90α与TGFβRI无相互作用。a)对质膜提取的蛋白进行HSP90α的免疫沉淀(IP),然后对TGFβRI和HSP90α进行免疫检测。IP与细胞外/膜蛋白先交联。IP作用于经rHSP90α处理或不经rHSP90α处理的IPF成纤维细胞48 h,作用温度为10 μM。IgG:非相关抗体;输入:非免疫沉淀提取物。erj - 00386 - 2017 - _figure_s5
图S6。IPF成纤维细胞中的HSP90α信号与TGF-β1通路无关。a) Western blot分析人原代对照成纤维细胞TGF-β1、α-SMA、p-ERK和ERK的表达情况。GAPDH染色作为加载对照。提出的WB具有3个独立实验的代表性。b)对照成纤维细胞经rHSP90α处理48 h (10 μM)和加入或不加入ALK5抑制剂SD-208 (30 μM)后,ELISA检测TGF-β1的总和活性水平。结果以平均值±SEM表示,**p<0.01, ***p<0.001, n = 5。c)对照成纤维细胞经rHSP90α处理后,在10 μM条件下,在1 mM条件下加入或不加入HS-30,用ELISA法测定其总TGF-β1水平和活性TGF-β1水平,结果表示为平均值±SEM, **p<0.01, ***p<0.001, n = 5。erj - 00386 - 2017 - _figure_s6
披露的信息
确认
作者要感谢马拉路德维希(麦吉尔大学,蒙特利尔,ON,加拿大)在组织浴设备设计方面的帮助。
脚注
本文的补充资料可从www.qdcxjkg.com
支持声明:C. Shimbori由肺纤维化基金会(I.M. Rosenzweig初级研究者奖)资助。P-S。Bellaye由le Fonds de Dotation " Recherche en Santé respiratory "和de la Foundation du Souffle、加拿大卫生研究院和加拿大肺纤维化基金会资助。本文的资助信息已存入Crossref基金管理人登记处.
利益冲突:可以在本文旁边找到披露www.qdcxjkg.com
- 收到了2017年2月23日。
- 接受2017年11月7日。
- 版权所有©ERS 2018











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}