





摘要
尽管许多慢性阻塞性肺病患者中存在血液或痰中嗜酸性粒细胞升高,但肺浸润嗜酸性粒细胞的解剖分布模式仍不确定。在慢性阻塞性肺病中,嗜碱性粒细胞几乎一直未被研究。本研究揭示了慢性阻塞性肺疾病肺组织浸润性嗜酸性粒细胞、嗜碱性粒细胞和嗜酸性粒细胞促进的免疫机制。
来自主要解剖部位的外科肺组织和活组织检查来自慢性阻塞性肺疾病全球倡议I-IV期严重程度分级的COPD患者;从不吸烟者/吸烟者作为对照。自动化免疫组织化学和原位杂交鉴定的免疫细胞,2型免疫标记GATA3和兴汀(CCL11,CCL24)。
嗜酸性粒细胞和嗜碱性粒细胞出现在受COPD受影响的肺部的所有解剖学中,并且在非常严重的COPD中显着增加。嗜酸性粒细胞症是惊人的斑块,富含嗜酸性粒细胞的微环境在空间上与GATA3连接+细胞,包括2型辅助T细胞淋巴细胞和2型先天淋巴细胞。在流感感染的小鼠中证明了类似局部化和白细胞介素-33 / ST2依赖性嗜酸性粒细胞症。小鼠和患者均用CCL11展示空间局限于狭窄的肠漾签名+成纤维细胞和CCL24+巨噬细胞。
除了识别组织嗜碱性作为晚期COPD一个新颖的特征,空间受限丰富嗜酸性粒细胞型2的微环境的识别表示在COPD的免疫病理学即可能对个性化治疗的影响的新颖类型的异质性。
摘要
在Copd受影响的肺部中鉴定了高度局部的TH2-和嗜酸性粒细胞的口袋,其数量增加,疾病严重程度增加并包括嗜碱性粒细胞。这举例说明了COPD免疫病理学中的新型异质性。http://bit.ly/2HexTco
介绍
慢性阻塞性肺病对全球发病率和死亡率的影响[1].这种疾病是由于长期接触吸入刺激物而引起的导致支气管炎、细支气管炎和肺气肿的慢性炎症(例如烟草烟雾)[2,3.].COPD病理学传统上归因于天生的免疫机制[3.,但适应性免疫机制也被激活[4,5].免疫病理学是由粒剖面的显着异质性进一步复杂化,具有增加的注意嗜酸性粒细胞中的签名COPD [6,7].多项研究表明,在相当大比例的COPD患者中,血液和/或痰中嗜酸性粒细胞计数高[6- - - - - -8].对痰粒细胞谱的聚类分析提出,富含嗜酸性粒细胞的痰嗜酸性粒细胞增多症是明显嗜酸性粒细胞COPD表型的标志[9].
2型细胞因子,特别是白细胞介素(IL)-5,对嗜酸性粒细胞的发育、成熟和组织寿命至关重要[10,11].这种IL-5依赖是通过中和抗IL-5 (mepolizumab)和抗IL-5受体-α (IL-5Rα;benralizumab)抗体(12,13]嗜碱性粒细胞也表达IL-5Rα,并受到IL-5/IL-5Rα阻断的影响[14].然而,关于copd患者肺组织浸润模式和嗜碱性粒细胞密度的资料有限。
慢性阻塞性肺病中肺组织浸润性嗜酸性粒细胞仍未被广泛研究。先前的研究已经证实在中央和远端室中存在嗜酸性粒细胞[15,16].然而,关于慢性阻塞性肺疾病中嗜酸性粒细胞增多的解剖定位和浸润模式以及其免疫触发机制的理论基础仍然存在重要问题。尽管2型细胞因子通常来源于抗原激活的CD4+2型辅助t细胞(Th2)淋巴细胞,嗜酸性粒细胞增多症可能通过激活的2型先天淋巴样细胞(ILC2)发展[17- - - - - -19],激活后(例如通过IL-33)可以快速安装瞬态2型应答[17,18].然而,如果ILC2细胞存在于受COPD受影响的肺部的嗜酸性粒细胞中的组织中,则仍有待证明。COPD中的Eosinophil Chemattractant分子也需要调查。
本研究的目的是对慢性阻塞性肺病(COPD)患者不同疾病阶段肺部组织浸润性嗜酸性粒细胞进行整体空间定位,并确定促进组织嗜酸性粒细胞增多的关键免疫学机制。研究涉及关键的解剖肺区,包括研究较少的远端肺区。此外,我们首次对慢性阻塞性肺病患者肺部浸润性嗜碱性粒细胞进行了系统量化。最后,利用一个实验模型,我们探讨了呼吸道病毒感染作为一种暂时性和斑片状嗜酸性粒细胞增多的潜在触发因素。
材料和方法
这里总结了材料和方法;更详细的程序载于补充材料.
从参与者的样品
对于主要研究,从Skåne大学医院(瑞典隆德,瑞典)的57名患者收集手术组织,并为组织学分析加工。从轻度到严重COPD的患者获得肺切除样品(慢性阻塞性肺病(金)阶段I-III的全球倡议,对照组(Never-Smokers /吸烟者)接受划定肿瘤的手术)。对于具有非常严重的(金IV)COPD的患者,移植手术后从外植入肺部获得肺组织。患者人口统计学呈现表1.mRNA-preserved组织为原位从30名患者的支气管活组织检查中收集杂交(ISH)(补充表E2).所有患者组的组织处理方案均相同。所有临床程序均由瑞典隆德当地的瑞典研究伦理委员会批准。所有参与者都签署了知情同意书。
免疫组织化学
抗原检索后(PT-LINK; Dakocytomation,Glostrup,Denmark),组织切片进行自动化的双重和三重免疫染色(AutoStainer Plus; Dakocytomation)。主要分析使用三重免疫组织化学(IHC)方案同时检测嗜酸性粒细胞,嗜碱性粒细胞和TH2替代标记GATA3。将Th2淋巴细胞被鉴定为GATA3+,CD4+细胞。ILC2细胞被鉴定为谱系阴性,CD25+,GATA3+混杂非ILC2细胞后细胞通过物理现有生色染色阻塞和阻断变性,如先前描述的20.].此外,各种标准IHC细胞标记物用于一般免疫细胞探测和鉴定趋化因子表达细胞。所有抗体已广泛验证用于临床诊断或研究(补充表E1).
现场杂交
使用RNAscope 2.5试剂盒(Advanced Cell Diagnostics, Hayward, CA, USA)观察人和小鼠eotaxin-1 (CCL11)和eotaxin-2 (CCL24) mRNA。切片用内源性酶块孵育,预处理缓冲液煮沸,靶探针杂交前用蛋白酶处理(CCL11, CCL24)。用显色原扩增靶RNA并显示。
定量IHC和计算机化图像分析
使用ScanScope幻灯片扫描仪(Aperio Technologies, Vista, CA, USA)对全部IHC和ish染色切片进行数字化。对高分辨率图像进行计算机分析,免疫反应性/检测色原进行颜色分割、量化并归一化到分析的组织区域(Visiomorph;Visiopharm Hørsholm、丹麦)。在主要研究中,对每位患者分析了包含细支气管、支气管(仅GOLD IV)、肺血管和肺泡实质的几个大切片,代表两到三个独立的肺区域。计算机分割的单个细胞的X,Y坐标通过空间分布和细胞聚集(点模式分析)以及在界定的嗜酸性粒细胞微环境中GATA3细胞的聚集(邻居分析,cell Community Viewer, CCV 1.22;瑞典隆德的Medetect和蒙特卡罗模拟;补充图E1).
胃癌和烟雾曝光的病毒恶化模型
保存肺鼠组织[21,22在暴露于病毒6天后终止的野生型或il -33缺陷小鼠(小鼠适应H1N1流感A;A/FM/1/47-MA),之前是否有11天每天接触烟草烟雾。这些研究得到了阿斯利康动物护理和使用委员会的批准。
统计分析
数据在GraphPad Prism (GraphPad Software, San Diego, CA, USA)中进行分析。非参数Kruskal-Wallis检验检测患者组之间的总体差异,Dunn检验进行评估事后群体间的比较保守。Spearman秩相关检验发现组间有显著相关。空间相互作用和二元点格局分析采用计算机点格局生成和圆形邻域分析(CCV;通过蒙特卡罗模拟测试(ImageJ v1.51S,美国国立卫生研究院,Bethesda, MD)和MOSAIC交互分析插件。在小鼠实验中,未配对t检验检测了病毒感染小鼠组之间的差异。Pearson参数相关和线性回归分析eotaxin mRNA与组织病毒含量的相关性。
结果
附加的结果在补充资料(表E2,图E1-E4).
在所有主要解剖肺部地区发生COPD中嗜酸性粒细胞和嗜碱性粒细胞的组织浸润
开展航空公司
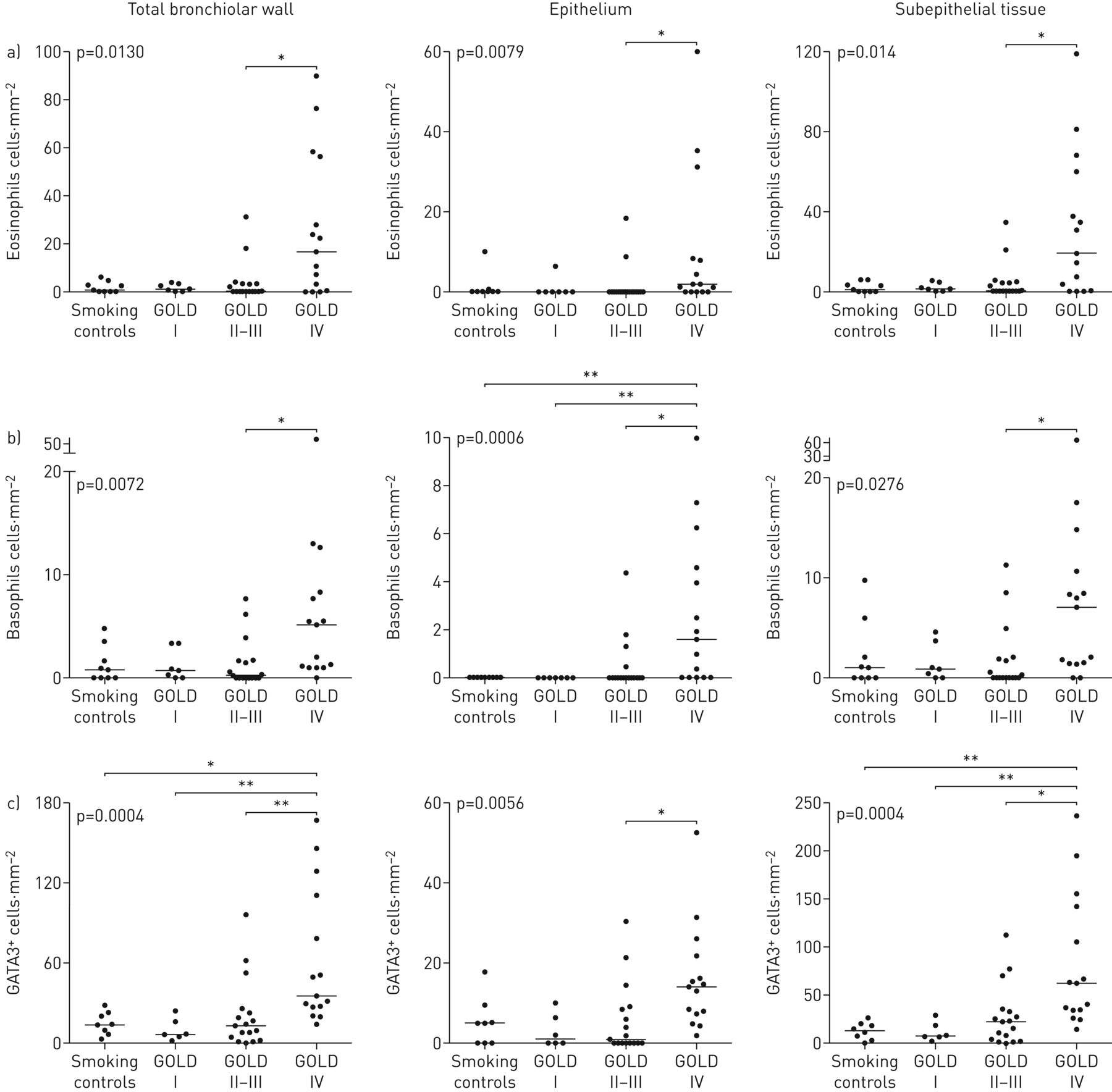
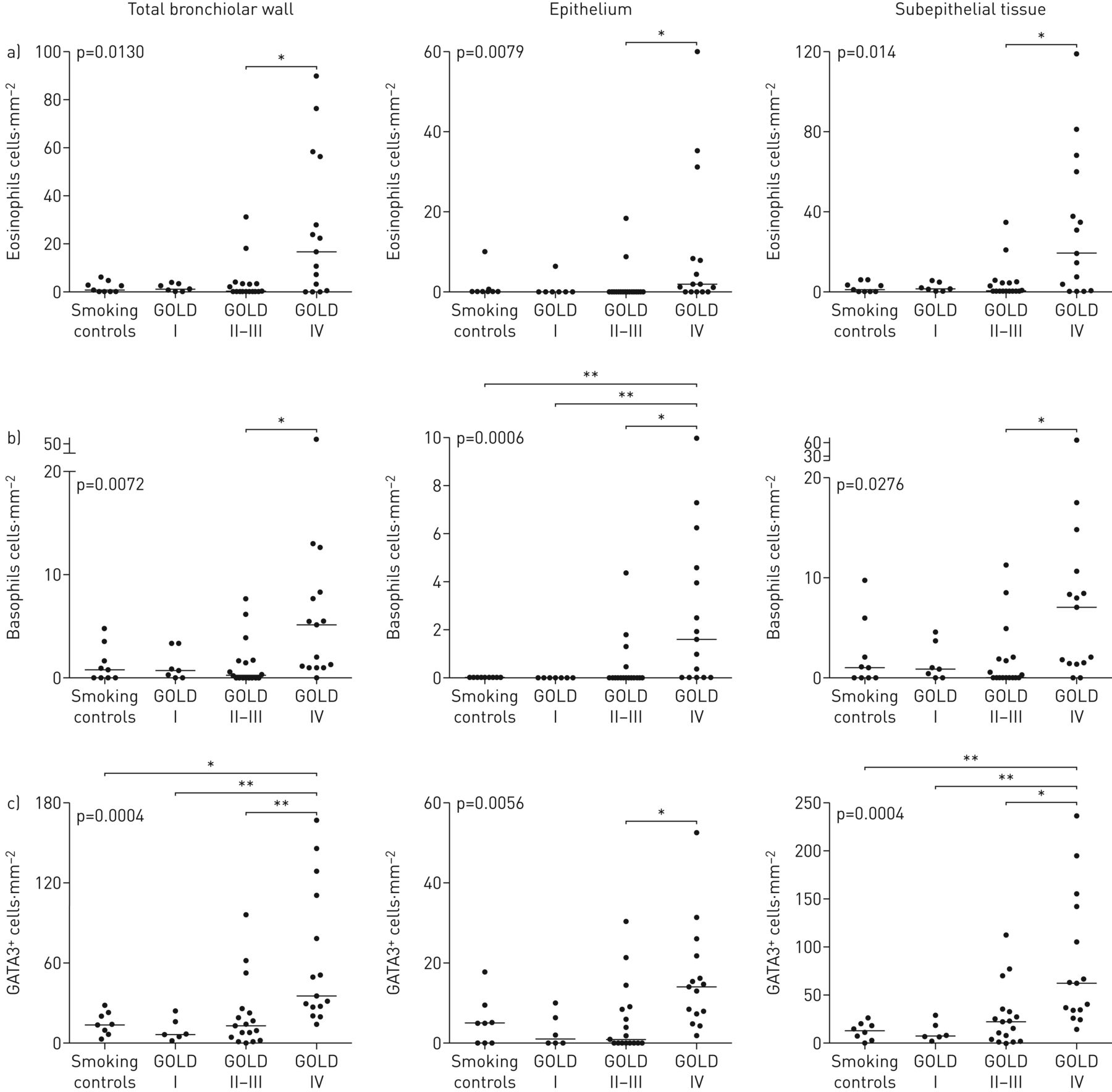
IHC在大多数患者中揭示了分散的支气管嗜酸性粒细胞和嗜碱性粒细胞。然而,与对照组(金IV)COPD(Gold IV)COPD(Gold IV)COPD(Gold IV)COPD(图1和2)嗜碱性粒细胞计数虽然小于嗜酸性粒细胞计数,但在晚期COPD中显著增加(图1).有趣的上皮嗜碱性粒细胞在对照和轻度疾病中缺乏缺乏缺乏患者患有Gold II-III的趋势,并且在黄金IV患者中显着上调。类似地,在晚期疾病中增加了2型替代标记物GATA3的密度(图1C.).同样GOLD IV患者的支气管(大气道)表现出与细支气管相似的高嗜酸性粒细胞、嗜碱性粒细胞和GATA3浓度(补充图E2C).在活检队列中,与不吸烟者相比,GOLD I-III期合并患者的嗜酸性粒细胞和嗜碱性粒细胞均有统计学上的增加,但不包括吸烟对照组(补充图E2A和B.).
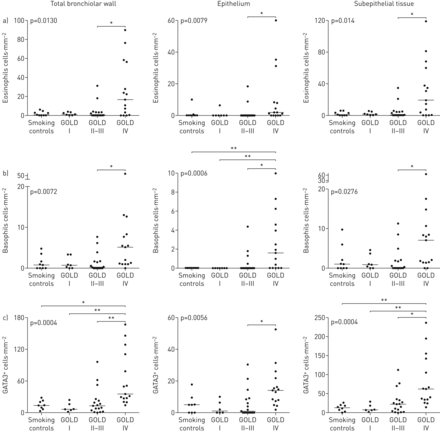
散点图显示慢性阻塞性肺病全球倡议(GOLD)患者细支气管(小气道)总壁、上皮和上皮下隔室中浸润的组织密度a)嗜酸性粒细胞和b)嗜碱性粒细胞和c)替代型2型免疫标记物GATA3I–IV期COPD和匹配对照组。数据以患者平均密度和组中值表示。图中引用的p值代表COPD患者和对照组之间的总体统计差异,由非参数Kruskal–Wallis单因素方差分析和Dunn多重比较确定事后测试(将每个子组的平均等级与每个其他子组进行比较)。*:P <0.05,**:P <0.01。
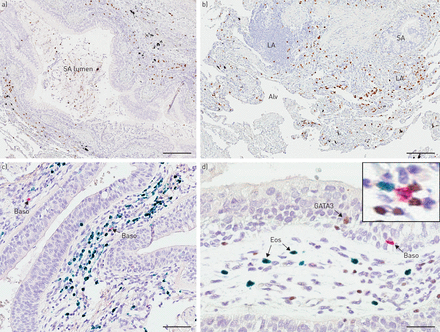
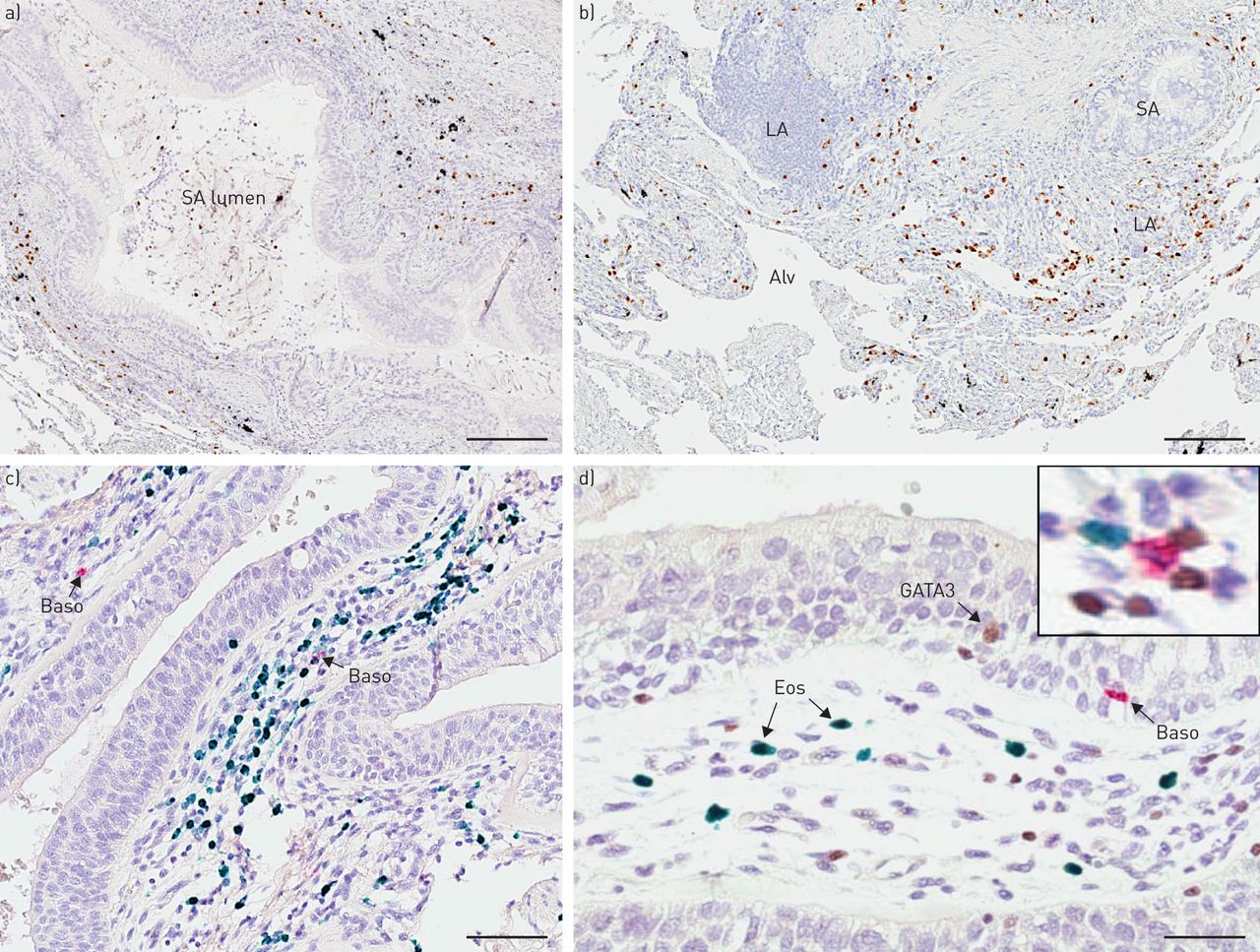
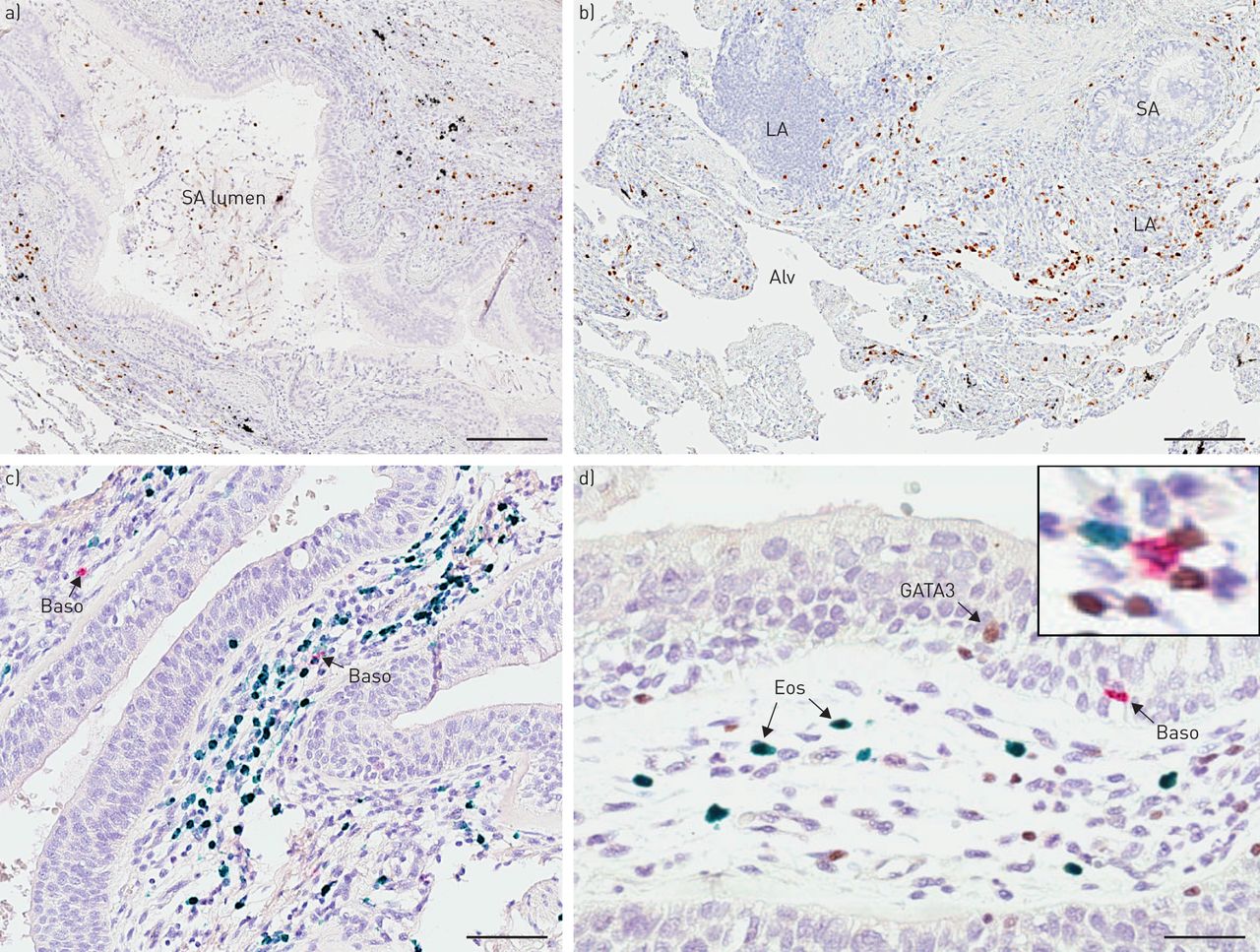
明亮视野显微镜显示慢性阻塞性肺病患者中嗜酸性粒细胞、嗜碱性粒细胞和GATA3染色,代表嗜酸性粒细胞高表达。a和b)单一嗜酸性粒细胞染色(棕色EG2免疫反应性);c)双免疫组化(IHC)肺切片染色,嗜碱性粒细胞(红色,碱性磷酸酶)和嗜酸性粒细胞(葡萄绿色)。d)嗜酸性粒细胞(绿色)、嗜碱性粒细胞(红色,碱性磷酸酶)和GATA3(棕色二氨基联苯胺;d插图为三染色切片和棕色GATA3的放大图+绿色EG2细胞+嗜酸性粒细胞和红色BB1+嗜碱细胞)。箭头表示阳性细胞。山:小气道;洛杉矶:淋巴聚集;Alv:肺泡薄壁组织;贝索:嗜碱细胞;Eos:嗜酸性粒细胞。规模的酒吧)100μm, b) 120μm, c) 85μm, d) 50μm。
远端肺隔间
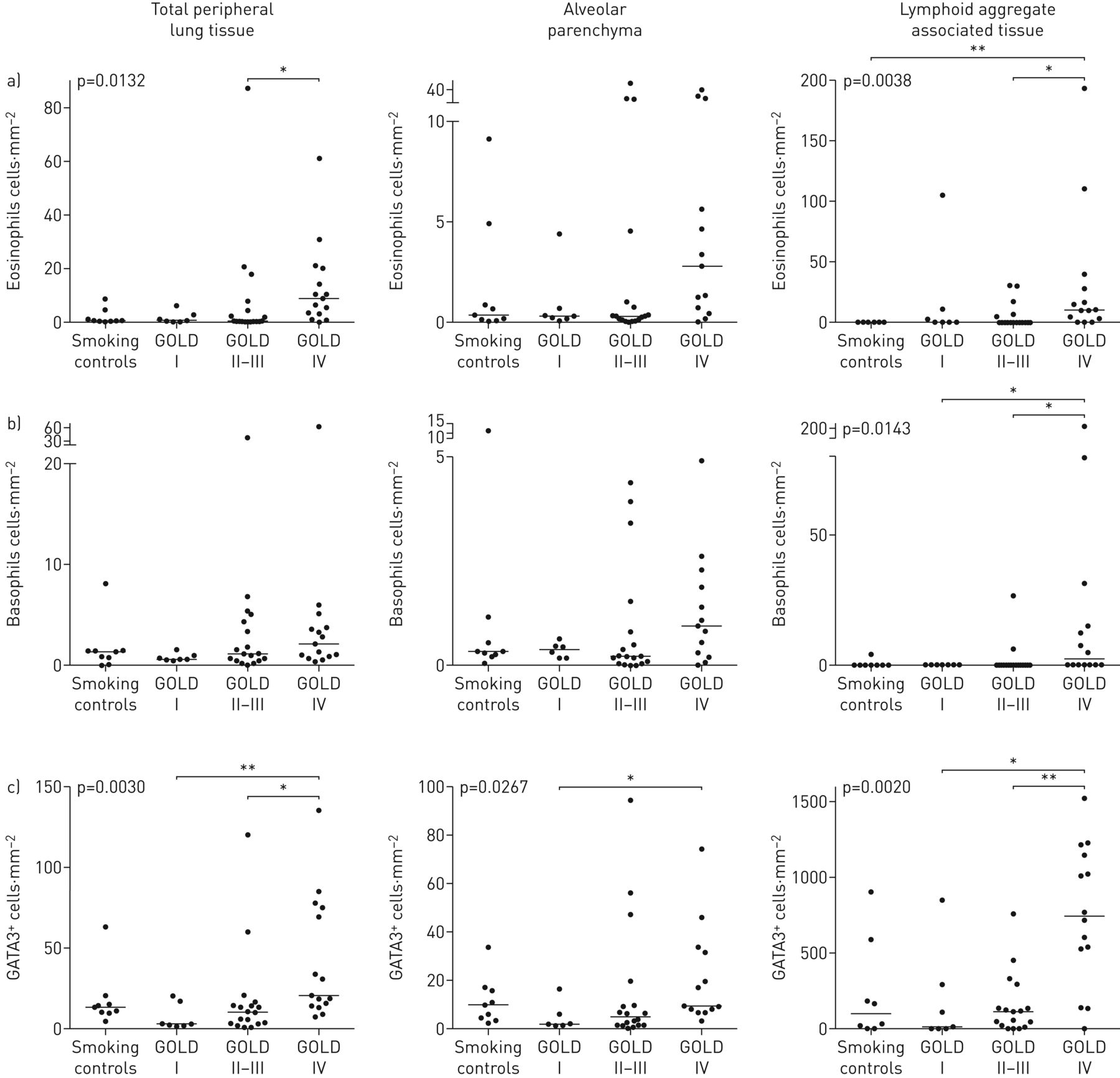
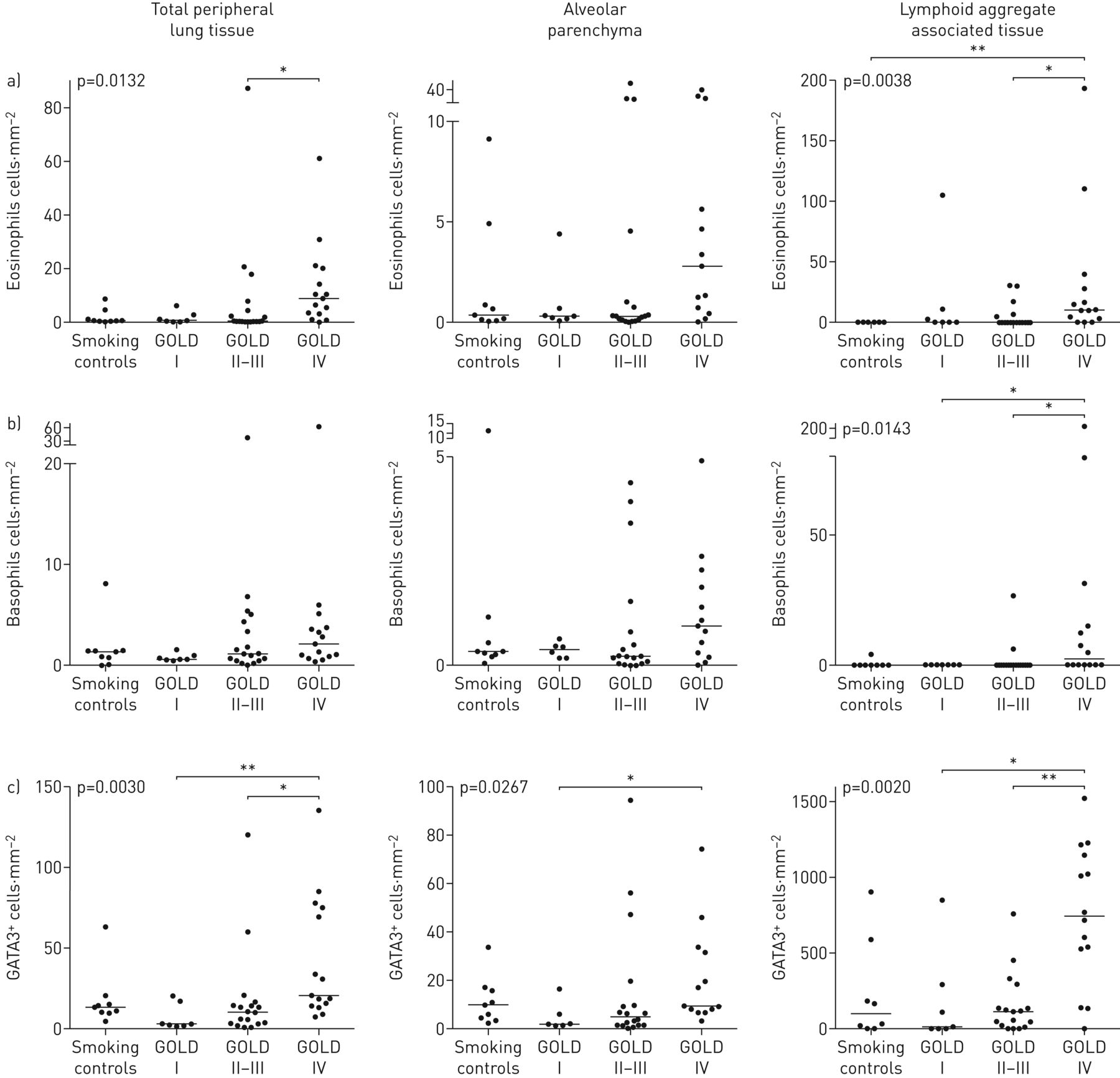
大多数患者在外围肺组织嗜酸性粒细胞可检测的浓度包括肺泡实质和远端淋巴组织。在GOLD IV期,观察到统计学上升高嗜酸性粒细胞的浓度(图3).特别高的嗜酸性粒细胞计数与异位淋巴滤泡(COPD的免疫病理学标志相关联;图2 b3a)也富含GATA3+细胞(图3C.).嗜酸性粒细胞计数越大,但与非COPD对照相比,嗜碱性粒子在远端肺隔室中显示出相对增加的密度相对增加的密度(图2.和3.).
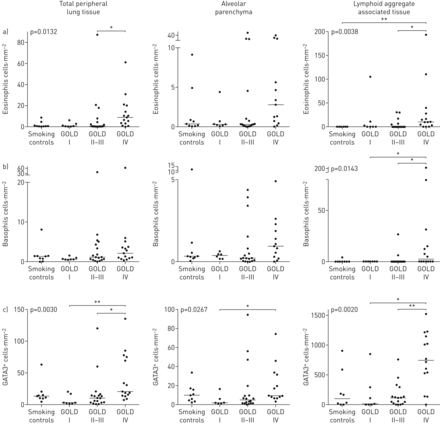
组织浸润a)嗜酸性粒细胞,b)嗜碱性粒细胞,c)远端肺室中替代性2型辅助t细胞标记物GATA3的密度散射图,这里分为整个外周肺组织(IE。排除大导管和大肺血管的肺组织)、肺泡实质(排除小气道、大到中等大小的血管和淋巴组织)数据以患者平均密度和组中位数表示。p值代表COPD患者和对照组之间的总体统计差异,由非参数Kruskal–Wallis单因素方差分析和Dunn多重比较确定事后测试(将每个子组的平均等级与每个其他子组进行比较)。黄金:慢性阻塞性肺病的全球倡议。*:P <0.05,**:P <0.01。
组织嗜酸性粒细胞和嗜碱性细胞浸润在空间上是相互联系的
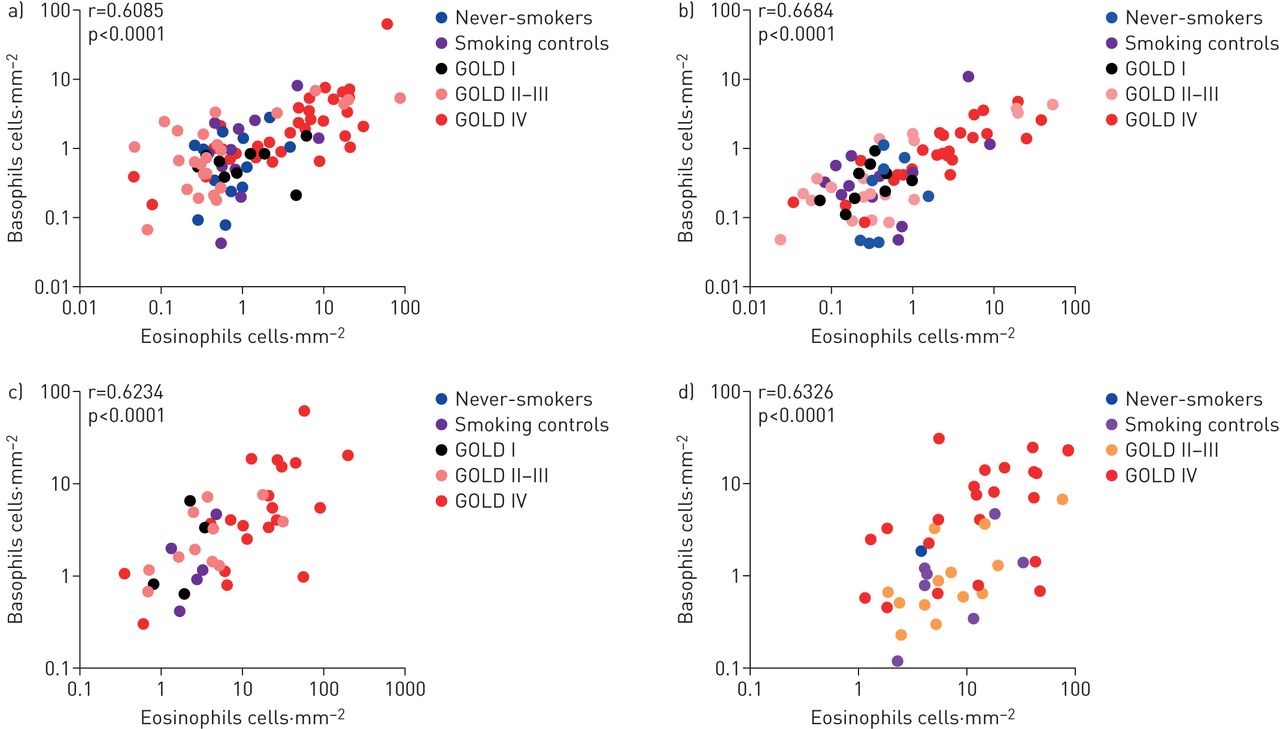
在同一组织环境中的嗜酸性粒细胞和嗜碱性粒细胞密度之间发现了显着的统计相关,这里由2-4厘米表示2手术切除。这种相关性存在于中央气道(支气管)和远端肺室(如小气道(细支气管)),整个周围肺组织和肺泡实质(图4).
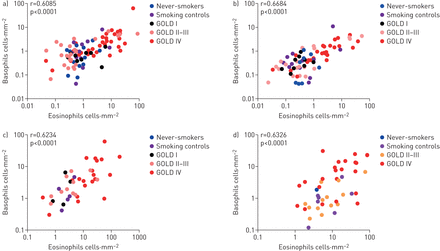
肺室嗜酸性粒细胞和嗜碱性粒细胞相关性散点图a)周围组织;b)肺泡薄壁组织;c)小航空公司;d)支气管。数据以单个组织块的平均值表示(IE。在每个分析的肺隔室内表示不同和空间分离的解剖区域的块)。值对数转换为产生相关性的更好可视化(作为省略任何单元类型的零值的结果部分)。虽然A-C代表主要研究的手术病例,D代表来自收集的金I-II患者的慢性阻塞性肺病(金)阶段IV阶段IV COPD样品和胚胎上海活组织检查的汇总平均值。收集的患者,得到mRNA保存的组织样品用于趋化因子mRNA可视化原位杂交。用Spearman秩相关检验确定相关程度。
COPD中的组织渗透嗜酸性粒细胞的积聚是斑块的,并且在空间不同的TH2微环境中浓缩
COPD肺部的不同嗜酸性粒细胞和嗜碱性细胞素微环境
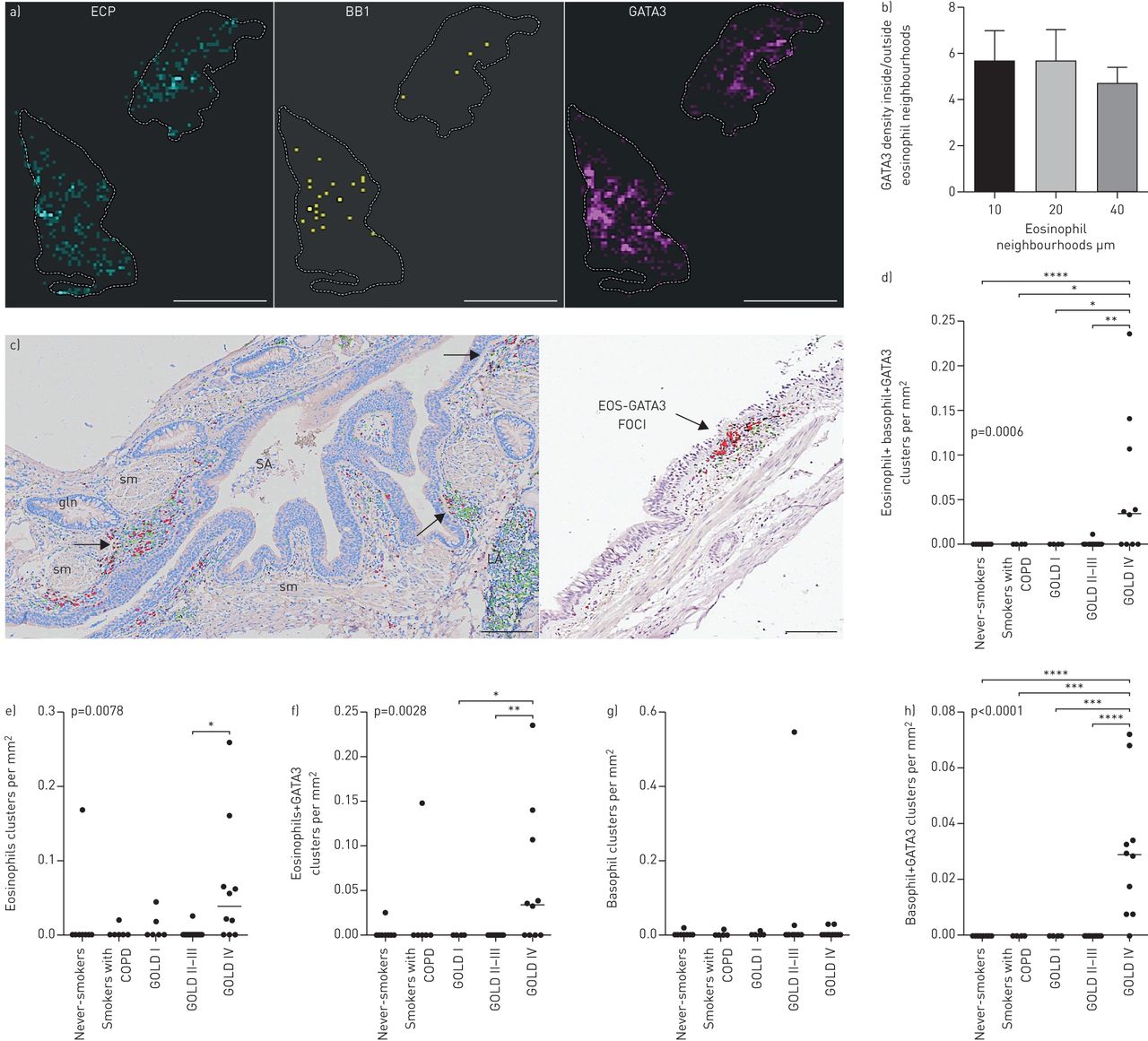
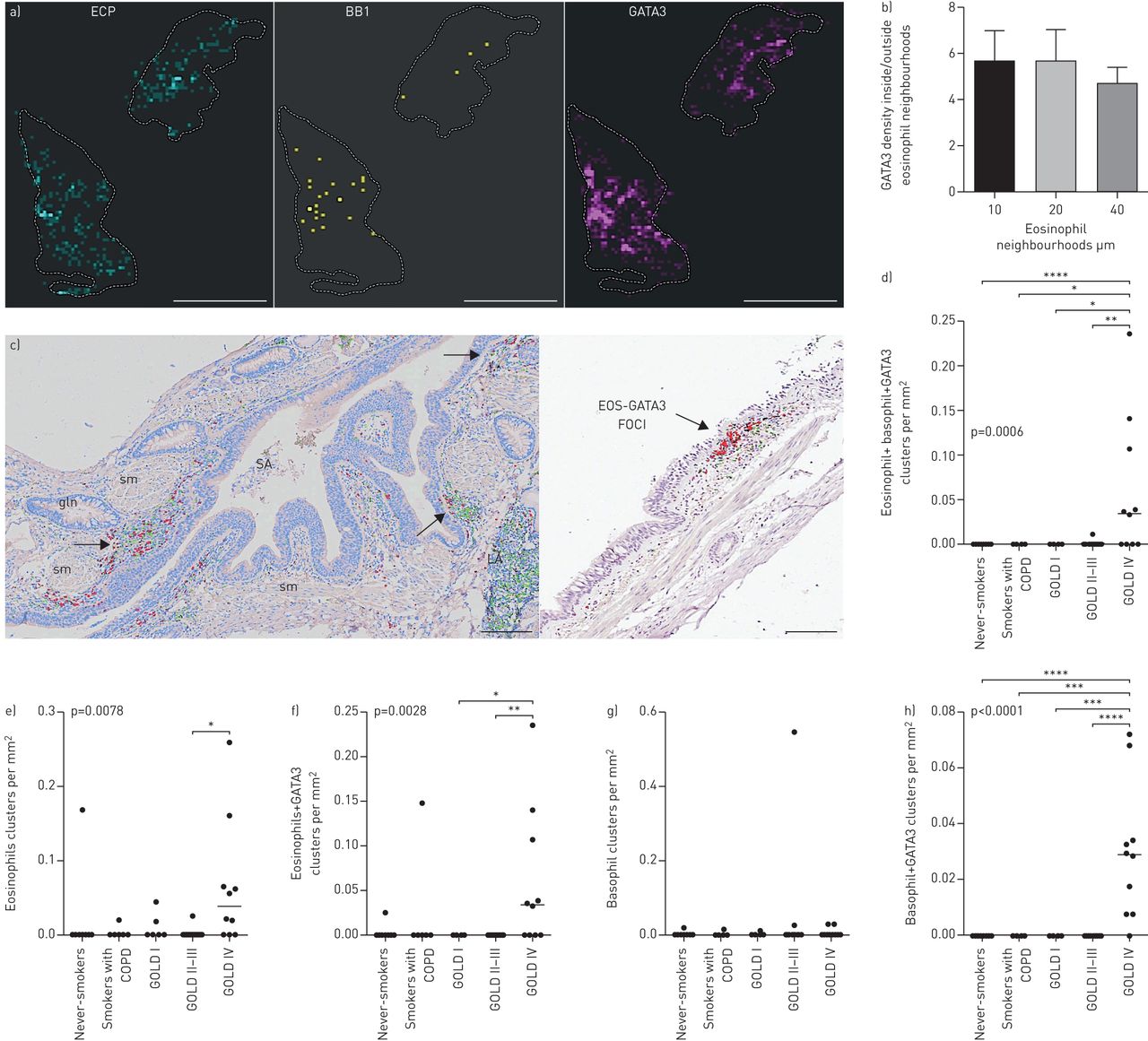
我们观察到微环境水平的嗜酸性粒细胞分布的显着斑块(图5)典型肺组织切片中的嗜酸性粒细胞通常局限于不同的局部微环境(嗜酸性粒细胞袋;图5a和c,补充图E1和E3).嗜酸性粒细胞和嗜碱性粒细胞簇的计数及其GATA3含量+在远端肺部块揭示了金IV COPD的显着簇增加(图5d).在所有解剖的肺区域都发现了空间嗜酸性粒细胞和嗜碱性粒细胞簇,最明显的例子是在支气管和细支气管固有层区域(小气道;邻居分析证实了统计学上的安全聚类,p<0.001)。
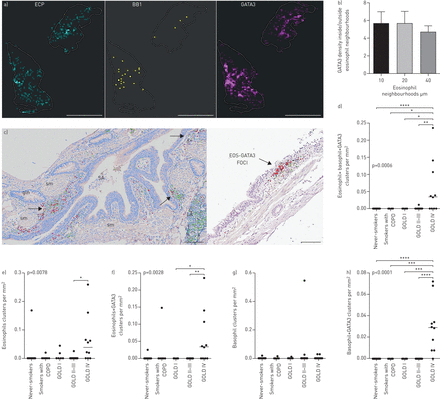
异构空间分布和COPD肺不同富嗜酸性粒细胞2型偏斜微环境的存在。一)空间连接热映射举例EG2的毛codistribution+嗜酸性粒细胞,BB1组+嗜碱性菌和GATA3,并描绘单个300×300 μm微环境,颜色编码为细胞密度(黑亮表示低-高密度)。b)空间统计分析(见补充图E1和方法细节文本)显示了嗜酸性粒细胞邻域内GATA3密度与嗜酸性粒细胞邻域外GATA3密度的商。显示了计算机创建的三个级别的圆形嗜酸性粒细胞邻域/微环境的数据,由半径10、20和40定义 单个嗜酸性粒细胞周围μm。c)嗜酸性粒细胞的明显空间病灶(计算机图像分析后伪彩色编码为红色)和GATA3(绿色)。嗜酸性粒细胞的聚集也通过点模式统计、最近邻距离分析和Ripley的K点模式分析得到证实(补充材料).d-h)含或不含任何GATA3细胞的嗜酸性粒细胞簇和/或嗜碱性粒细胞簇的组织密度的定量数据。数据显示为每毫米患者平均群集数2肺组织和组中值。p值代表COPD患者与对照组之间的总体统计差异,由非参数Kruskal-Wallis单因素方差分析与Dunn的多重比较确定事后测试(每个亚组的平均等级与其他亚组进行比较)。EOS:嗜酸性粒细胞;gln:上皮下腺;LA:淋巴聚集;SA:小气道;sm:平滑肌。标尺a)1 cm,b)150 μm,c)250 μm.*:p<0.05,***:p<0.01,***:p<0.001。
嗜酸性粒细胞微环境链接到本地化GATA3签名
反复染色进一步表明,富含嗜酸性粒细胞的微环境的存在伴随着嗜碱性粒细胞和GATA3的焦点聚集+细胞(图5a空间统计分析方法用于量化GATA3的密度+内部和外部的嗜酸性粒细胞微环境的细胞(补充图E1).对于斑片状嗜酸性粒细胞增多的肺样本,GATA3的密度和聚类+与非邻近区域相比,嗜酸性粒细胞邻近微环境中的细胞明显增加数倍(图5b).一种空间GATA3嗜酸性粒细胞的关系通过点图案Monte Carlo模拟(P <0.001)证实。
嗜酸性粒细胞微环境中ILC2和Th2淋巴细胞的存在
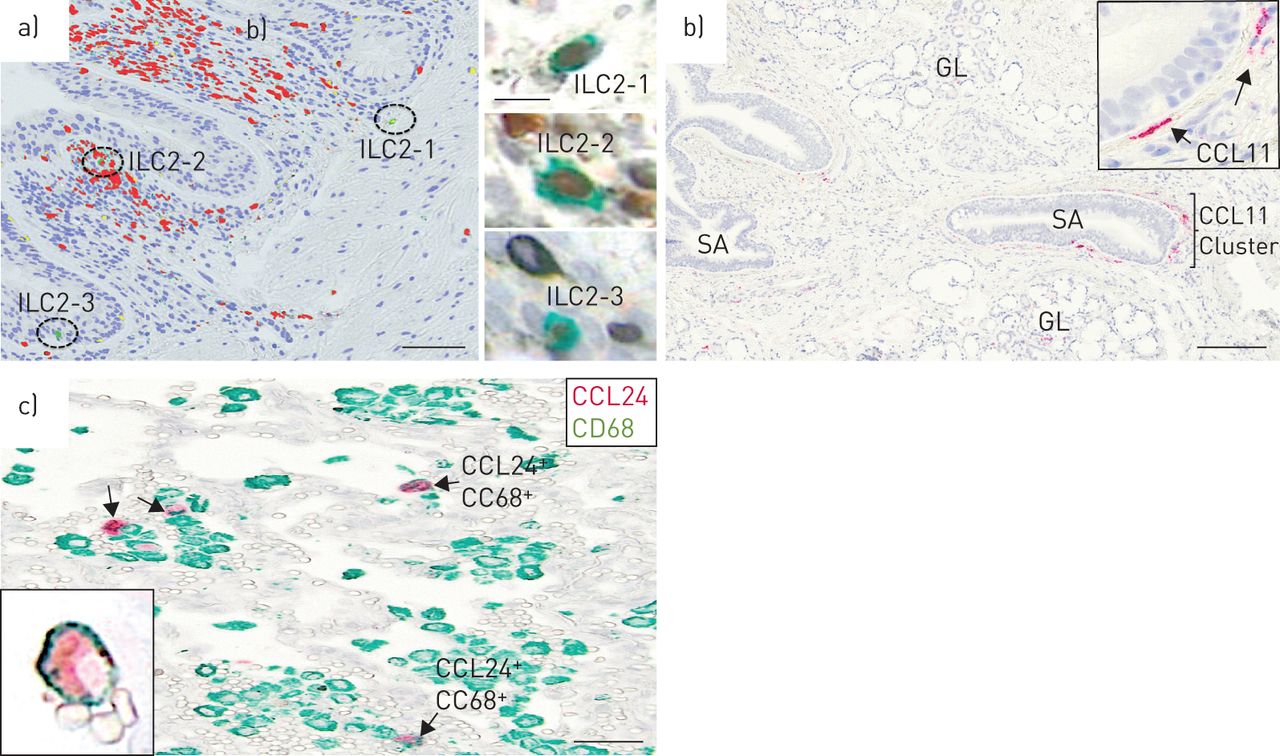
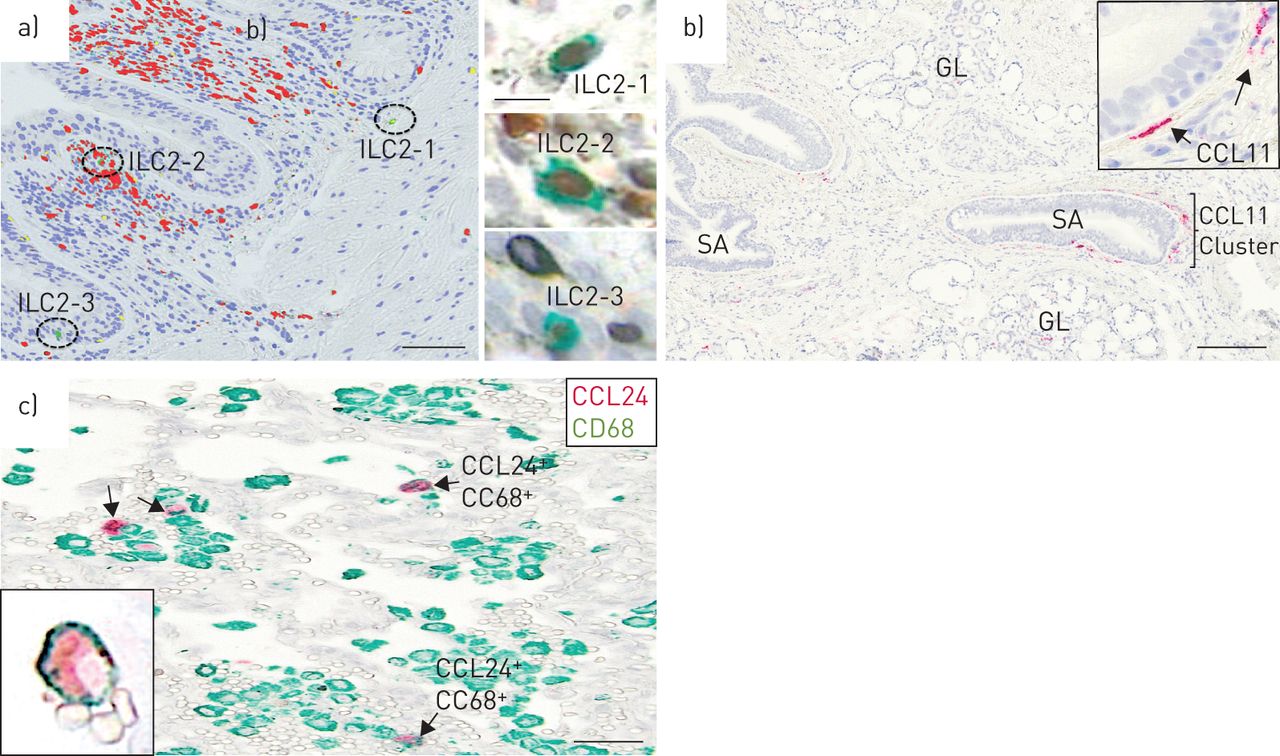
ILC2细胞可以代表局部2型细胞因子源,促进本型局部嗜酸性粒细胞瘤的局部类型。我们独特的ILC2染色协议表明嗜酸性粒细胞焦点内的ILC2细胞的清晰定位(图6a).正如预期的那样,ILC2计数明显低于经典CD4+Th2淋巴细胞与ILC2的ILC2分数的ILC2和嗜酸性粒细胞患者中的TH2细胞的分数为±sd3.5±3.8%。值得注意的是,气道上皮中的ILC2细胞的馏分是ILC2和TH2细胞总和的15.5±18%。
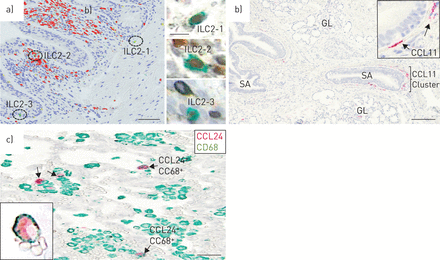
a)细支气管嗜酸性粒细胞(红色)灶和家族阴性GATA3存在的例子+CD25+慢性阻塞性肺疾病(GOLD) IV患者全球倡议中的2型先天淋巴样细胞(ILC2)(绿色;细胞核为蓝色)。插入物描绘了相同ILC2s的原始显微照片(GATA3棕色;CD25绿色)。b)亮场图像显示CCL-11 (eotaxin 1) mRNA表达的局灶分布。CCL-11 mRNA检测原位用永久性红色底物显色剂杂交和显示。低功耗概述说明了CCL-11的局部集群+而放大后(b插图)显示CCL-11呈细长的成纤维细胞样形态+细胞。c) cd68阴性CCL11例+(只有绿色)细胞和CD68+CCL24+(红色和绿色)细胞在受copd影响的肺中。GL:上皮下粘液腺;SA:小气道(IE。细支气管)。比例尺a) 100 μm(插图10 μm), b) 200 μm, c) 50 μm。
呼吸道病毒感染的勘探如片状和空间上不同的嗜酸性粒细胞积聚在肺组织的潜在原因
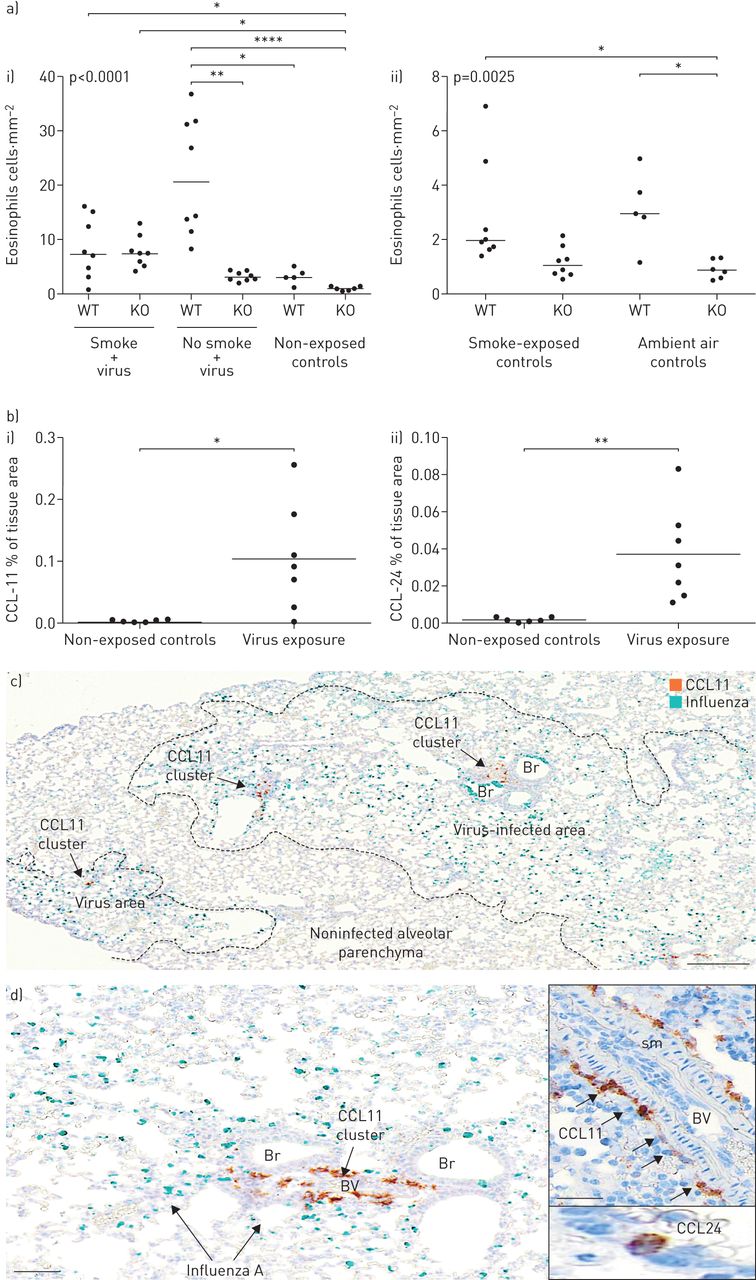
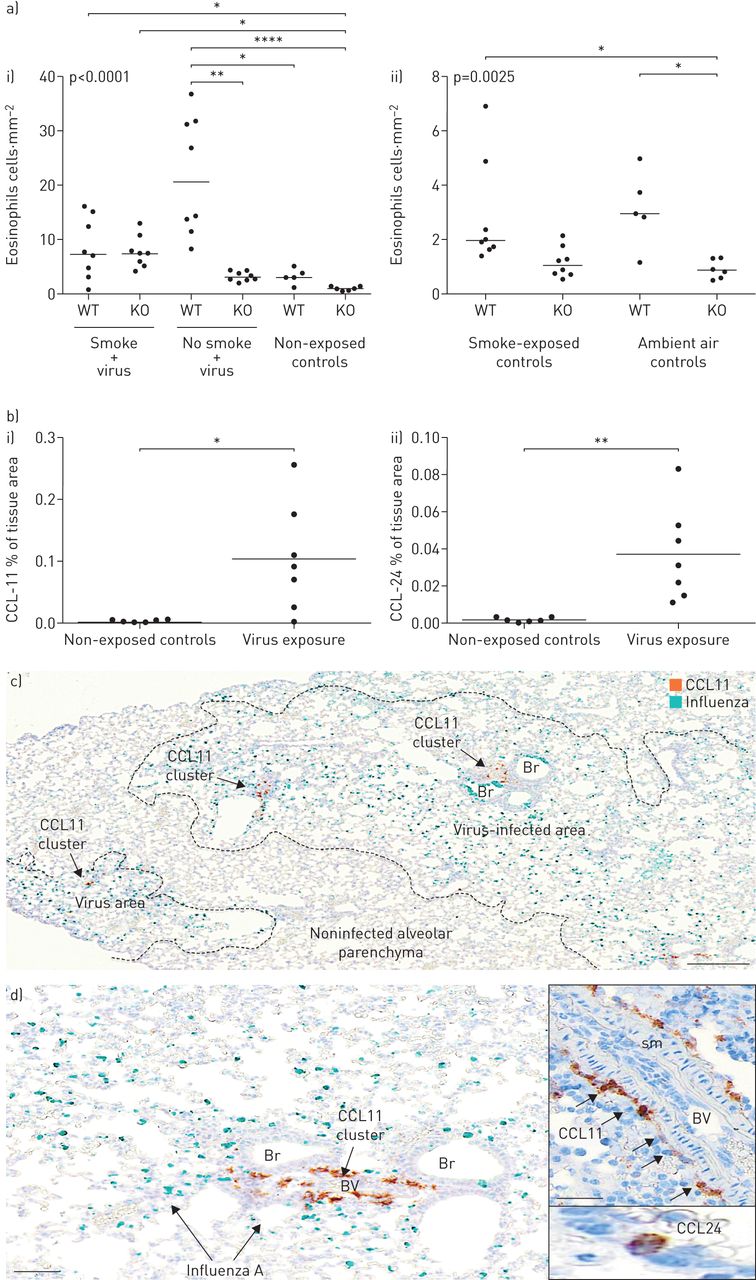
我们的研究结果,从烟雾和/或流感病毒感染的经验证小鼠恶化模型的肺组织中嗜酸性粒细胞响应探查,表明病毒感染引起嗜酸性粒细胞的浸润健壮(图7A和补充图E4d).生成的嗜酸性粒细胞差异是斑块状的,并将其上透明化为本模型中描述的斑块感染(流感存在)(图7) [21].小鼠暴露于现有烟草烟雾没有安装在感染了类似的嗜酸性粒细胞反应(图7).流感诱导的嗜酸性粒细胞增多似乎与IL-33/ST2轴相关,因为病毒诱导的嗜酸性粒细胞增多并不发生在IL-33中−−/老鼠(图7A)一世)。IL-33本身的敲除显着降低了无感染野生型小鼠中嗜酸性粒细胞的低级基线存在(图7A)(二)。
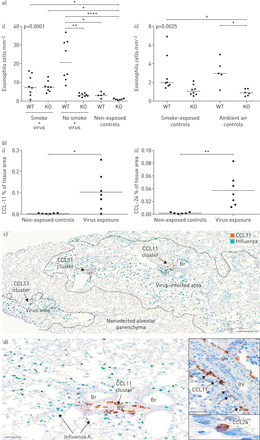
流感病毒感染触发了一种斑态嗜酸性粒细胞症和高嗜酸性趋化因子的高度局部诱导。a)感染诱导的嗜酸性粒细胞菌。在流感后6天,在6天内,在6天的情况下,在嗜血症后6天内呈现野生型(WT)和白细胞介素(IL)-33敲除(KO)小鼠I);ii)环境空气和烟草烟雾防止对照中嗜酸性粒细胞的无活性基线计数(无病毒)。数据显示为WT和IL-33 KO小鼠。b)肺组织I)CCl11和II)在感染后6天内对照和病毒感染的WT小鼠中的CCL24免疫反应性。C和D)感染后6天在小鼠肺部在小鼠肺部本地化CCL11的空间局部存在。病毒感染区域中典型和高度局部CCL11簇(棕色)的例子(病毒通过vina绿色色原染成病毒)。注意支气管和支气管相关肺血管(BV)周围的不同CCL11定位。插入d举例说明了血管内CCL11+细胞以及典型的孤立性CCL24+肺泡细胞。Br:支气管;sm:平滑肌。比例尺c) 200 μm, d) 100 μm(插图50 μm)。
copd感染的肺和病毒感染的小鼠显示eotaxins的局部表达
Eotaxins是嗜酸性粒细胞迁移和组织归巢的关键化学引诱剂。联合ISH+IHC用于揭示eotaxin表达细胞在COPD患者和病毒感染小鼠中的存在和身份。小鼠感染病毒后,总肺CCL11和CCL24均显著上调(图7B.).CCL11的表达很明显,局限于大的血管周结构细胞(图7C.和d)主要局限于感染和富含嗜酸性粒细胞的肺部区域(图7C.和d). CCL24的分布比CCL11更广泛,并且主要局限于受感染和富含嗜酸性粒细胞的肺区域的肺泡巨噬细胞(补充图E4c).
在人类慢性阻塞性肺病组织中,CCL11有一个可变的斑片状定位,并由一个细长的成纤维细胞样亚群(波形蛋白)表达+)结构的细胞。与小鼠相比,受copd影响的人肺中的CCL11主要位于气道或腺上皮下(图6b),也是嗜酸性粒细胞聚集最显著的部位。表达ccl24的细胞以CD68为主+肺泡巨噬细胞(图6c),且在视觉上没有明显的嗜酸性粒细胞簇共定位。
讨论
这项研究揭示了慢性阻塞性肺疾病中嗜酸性粒细胞、嗜碱性粒细胞和Th2免疫的几个新方面,特别是慢性阻塞性肺疾病影响的肺部缺乏炎症均匀性。该研究排除了有特应性反应或过敏史的患者。因此,这些数据揭示了COPD特异性嗜酸性粒细胞增多的本质,以及目前在COPD和非过敏性哮喘表型中正在密切研究的嗜酸性粒细胞增多型的Th2免疫。有趣的是,嗜酸性粒细胞增多的慢性阻塞性肺病已经被证实表现出嗜酸性粒细胞增多的哮喘的组织病理学特征,例如基底膜网状层增厚[23].这增加了共享基础类型2机制的可能性。因此,尽管我们在本研究中对COPD的诊断有信心,但我们承认其定义及其在非过敏性嗜酸性粒细胞哮喘合并固定梗阻、嗜酸性粒细胞增多COPD和哮喘- COPD重叠综合征之间的鉴别诊断的复杂性[24].
患有严重疾病的COPD患者接受大剂量吸入糖皮质激素治疗后,观察到嗜酸性粒细胞和嗜碱性粒细胞数量升高,这一事实进一步支持了对哮喘的观察,即非过敏性嗜酸性粒细胞增多症比经典的变应原诱导的嗜酸性粒细胞增多症更耐类固醇。此外,这也与COPD晚期患者肺泡一氧化氮浓度增加的报道一致,表明远端气道非过敏性2型信号增加[25].对COPD患者类固醇敏感的问题是通过在温和的疾病是增加的反应的指标的血液或痰嗜酸粒细胞增多的观察复杂[26,27].值得注意的是,在我们的子活检研究中,嗜酸性粒细胞和嗜碱性粒细胞也较温和的疾病增加。但是,因为没有完美的年龄和性别匹配对照组患者,这一结果应谨慎解读,应该指出的是,我们的研究样本量不允许对类固醇反应的任何结论。
先前研究发现嗜酸性粒细胞是在COPD患者的一个子集[一个显著特点6].在本研究中,仅在非常严重的COPD中检测到嗜酸性粒细胞症的统计增加。应该指出的是,所有黄金阶段IV患者都停止吸烟,而〜40%的升级患者是目前吸烟者。因此,因为烟雾暴露可以抵消型2型反应(其在这里由本小鼠实验支持),所以不能排除持续吸烟可能导致较低嗜睡性疾病中观察到的下嗜酸性粒细胞症。此外,目前的研究设计不能通过促进止血剂I-III患者发生的程度嗜酸性嗜酸性粒细胞。一种新的研究发现是解剖学上普遍的嗜酸性粒细胞渗透,涉及受COPD受影响的肺部的所有主要解剖室。结果补充了另一项研究报告,患有COPD和更高血液粒细胞计数的患者(> 250个细胞·μL−1)与血液嗜酸性粒细胞计数的患者相比,相应更大的痰和支气管肺泡灌洗嗜酸性粒细胞计数(<150个细胞·μl−1) [28].这些嗜酸性嗜酸性粒细胞患者还增加了各种肺部位置的嗜酸性粒细胞和更大的组织重塑[28].除了之前在非过敏性慢性阻塞性肺病患者中发现的腔内和支气管嗜酸性粒细胞增多[9,29,30.], COPD中的嗜酸性粒细胞可能在细支气管、肺泡实质和异位淋巴聚集物中发挥效应功能。
另一个主要的观察是,嗜酸性粒细胞在微环境水平上的分布呈明显的斑块状。这表明,非过敏性慢性阻塞性肺疾病患者的嗜酸性粒细胞增多的潜在原因(但尚未确定)可能来自于高度局部性的外部来源。这也意味着引发和表现嗜酸性粒细胞增多症的免疫反应可能也是高度局部性的。我们发现嗜酸性粒细胞和2型标记GATA3之间存在明确的空间关系,支持了这一观点。GATA3的表达对Th2淋巴细胞和ILC2细胞产生2型细胞因子至关重要[31- - - - - -33],并以在诊所对抗2型免疫为目标[30.].因此,重要的是发现不仅是GATA3+th2淋巴细胞,但也是gata3+ILC2细胞位于富含嗜酸性粒细胞的囊袋中。ILC2细胞具有产生2型细胞因子的能力,此前已在小(约0.05%的CD45)的COPD肺中得到证实+细胞),但可能是生物学相关的数字[34].同样,我们的研究显示Th2细胞在数量上明显优于ILC2s,尽管在上皮内层ILC2/Th2的比例仅为约1:6。
我们高度本地化的Th2和慢性阻塞性肺病,肺的影响富含嗜酸性粒细胞口袋的发现是概念新颖,并评估患者的炎症状态的一个重要的未来的挑战。例如,血液或痰嗜酸粒细胞增多指示组织嗜酸性粒细胞的存在,但并不排除一个显著部分或其他肺部区域将具有一种天生的巨噬细胞和嗜中性粒细胞富 - 或1型T辅助细胞类型炎症的可能性。这代表了个性化的改进治疗显著未来的挑战,并有助于了解指定类型的慢性阻塞性肺病肺2机制的挑战。
在达成的人和小鼠的出版研究中,表明病毒感染可以引起短暂的嗜酸性粒细胞[35- - - - - -37[本研究证实,在小鼠肺部流感感染后,鲁棒嗜酸性粒细胞显现。在我们的实验模型中,我们进一步揭示这种嗜酸性粒细胞反应是高度斑块的,与空间局部化的肠昔姻反应相关,并且依赖于IL-33释放,因为嗜酸性粒细胞在IL-33中没有发展−−/老鼠。此外,与实验性心肌炎模型一致[38,我们联合ISH和IHC发现CCL11主要由成纤维细胞和周细胞表达,而CCL24则定位于巨噬细胞。我们的实验数据,以及病毒感染在慢性阻塞性肺病恶化期间很常见的事实[39],将斑驳的局部感染作为本新型新型斑块2型蛋白质类型2,富含嗜酸性粒细胞的口袋的刺激性触发器。实际上,我们的数据表明,在晚期疾病中,斑态嗜酸性粒细胞更常见,在此期间,与早期阶段相比,患者发展更多的感染[39].然而,我们的研究的局限性是,患者群体相对较小,斑块性嗜酸性粒细胞增多症的感染原因仍然是间接的。因此,需要进行更大规模的研究,对微生物(细菌和病毒)进行更广泛的评估。
此外,这项研究首次对慢性阻塞性肺病组织浸润性嗜碱性粒细胞进行了系统的定位。嗜碱性粒细胞的密度与嗜酸性粒细胞的密度相关。然而,由于嗜碱性粒细胞在受控情况下实际上是不存在的,相对于通常观察到的许多其他免疫细胞的增加幅度要大得多。有趣的是,类似于对哮喘的观察[40,嗜碱性粒细胞的组织密度随疾病严重程度而增加。首选浸润部位为远端肺和传导气道的异位淋巴组织,例如,GOLD IV型患者小气道上皮内嗜碱性粒细胞显著增加。这种渗透模式与嗜碱性细胞作为有效免疫调节剂和宿主防御细胞的现代观点相一致[41,42].例如,嗜碱性粒细胞可通过IL-4分泌放大局部的2型反应[11,43],被IL-33和胸腺基质淋巴生成素激活,除组胺外还可能释放多种促炎细胞因子。在最近的实验模型中,嗜碱性粒细胞已被确定在肺气肿的发展中起作用[44它们还与对copd相关细菌和病毒的防御有关[41].因此,嗜碱性粒细胞除了具有有害的致病能力外,还可能发挥保护作用。无论如何,目前的研究表明,像嗜酸性粒细胞一样表达IL-5Rα的嗜碱性粒细胞应该被认为是IL-5-和IL-5Rα靶向治疗的潜在相关靶点。
COPD组织中的嗜酸性粒细胞在多大程度上发生碎片性脱颗粒、程序性细胞溶解或继发性坏死仍有待探索[43,45].支气管肺泡灌洗液或哮喘和COPD中的痰液样品的升高的自由颗粒蛋白经常被视为活性脱粒的标志[46]但是腔内颗粒蛋白的解放是由腔内坏死机制明显引起的。不幸的是,我们的样品不适合电子显微镜,防止超微结构分析脱粒状态,并且COPD组织中的嗜酸性粒细胞的活化状态也仍有待确定。
总而言之,这项研究确定了嗜碱性粒细胞在慢性阻塞性肺病中具有潜在作用,并证明慢性阻塞性肺病中的组织嗜酸性粒细胞在解剖学上广泛存在,但通常局限于明显的th2倾斜和ilc2包含的微环境。这种组织嗜酸性粒细胞增多的特征可能具有临床意义。此外,我们的数据表明,呼吸道感染是慢性阻塞性肺病斑块性嗜酸性粒细胞增多的潜在触发因素。
补充材料
可共享的PDF
致谢
我们感谢Karin Jansner (Medetect AB, Lund, Sweden)和Britt-Marie Nilsson (Lund University, Lund),感谢他们娴熟的组织准备和组织学工作。在作者的指导和指导下,由Debra Scates (JK Associates, Inc., Conshohocken, PA, USA)和Michael A. Nissen (AstraZeneca, Gaithersburg, MD, USA)提供编辑支持和手稿提交。这项支持是由阿斯利康公司资助的。
脚注
本文提供了补充材料www.qdcxjkg.com.
利益冲突:P. Jogdand没有什么可披露的。
利益冲突:P. Siddhuraj没有什么可披露的。
兴趣冲突:M. Mori没有什么可披露的。
利益冲突:C.桑德没有什么可以披露的。
利益冲突:J. Jönsson没有什么可披露的。
利益冲突:A.F.墙壁无需披露。
利益冲突:J. Kearley是Astrazeneca(前身Medimmune LLC)的雇员,并在Astrazeneca有股票期权。
利益冲突:A.A.在进行这些分析时,谦卑是Astrazeneca(以前Medimmune LLC)的员工。
利益冲突:R. Kolbeck在进行这些分析时是阿斯利康(前MedImmune LLC)的员工。
利益冲突:L. Bjermer没有什么可披露的。
利益冲突:P.纽博尔德是阿斯利康公司(前身为MedImmune公司LLC)的员工,并在阿斯利康的股票期权。
利益冲突:J.S. Erjefält是Medetect AB的创始人(和股票持有人),他获得了阿斯利康的资助,进行了本研究的部分内容。
支持声明:本研究的资金由AstraZeneca,瑞典心脏和肺基金会和瑞典研究委员会提供。本文的资金信息已存入CrossRef Resder注册表.
- 收到了2019年1月15日。
- 接受2020年2月6日。
- 版权©2020人队
此版本在Creative Commons归因非商业许可证4.0的条款下分发。








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}