


文摘
Guanylyl环化酶是一个家族的酶催化三磷酸鸟苷,cGMP的转换。家庭由膜结合和可溶性亚型,都表示在几乎所有类型的细胞。他们是由不同的细胞外受体激动剂包括肽激素、细菌毒素、自由基,以及细胞内的分子,如钙和腺嘌呤核苷酸。刺激guanylyl环化酶的合成积累cGMP调节复杂的信号级联通过直接下游效应器,包括cGMP-dependent蛋白质激酶,cGMP-regulated磷酸二酯酶,和循环nucleotide-gated离子通道。Guanylyl环化酶和cGMP-mediated信号级联发挥核心作用在不同的生理过程(patho)的规定,包括血管平滑肌蠕动肠道液体和电解质内稳态,视网膜phototransduction。解决综述的主题包括结构和基因的染色体定位guanylyl环化酶,结构和功能的guanylyl酸环化酶家族的成员,分子机制调节酶活性,催化活性和配体分子序列耦合绑定。简要概述了下游事件由guanylyl控制环化酶,包括由cGMP的感受器和角色guanylyl环化酶在细胞生理和病理生理学。
我的介绍。
Guanylyl环化酶进化合成cGMP,以应对不同的信号,如一氧化氮(NO)、2多肽配体,在细胞内Ca和通量2 +([Ca2 +]我)。这些信号使用特定guanylyl cyclase-coupled受体和代数余子式启动转换的胞质嘌呤核苷三磷酸鸟苷环鸟苷酸。细胞内cGMP ((cGMP)我)调节细胞生理通过激活蛋白激酶,直接控制特定的离子通道,或改变细胞内的环核苷酸磷酸二酯酶的浓度通过监管(pd)。家庭的结构和功能的guanylyl环化酶,调节他们的活动的分子机制,和下游效应器cGMP-dependent的生理学过程总结综述。
二世。Guanylyl环化酶
答:分子生物学
1。Guanylyl酸环化酶家族的成员的识别。
成立的1970年代中期guanylyl环化酶活性被发现在大多数细胞的可溶性和颗粒分数(Hardman萨瑟兰,1969;石川et al ., 1969;舒尔茨et al ., 1969;Aurbach白色和,1969年),这些活动是由不同的蛋白质(加伯和灰色,1974年;木村的Murad, 1974;克里斯曼et al ., 1975)。然而,只有随着分子克隆技术的发展超过十年之后这种酶家族的宽度可以充分探索(表1和2)。净化guanylyl酸环化酶的胞质间透露了可溶性同种型α-β-subunits组成的异质二聚体。β-subunit有分子质量∼70 kDa,而α-subunit报道是73到82 kDa (Gerzer et al ., 1981 c;Kamisaki et al ., 1986)。可溶性guanylyl环化酶(国网公司)纯化从牛或鼠肺(明显的同质性Koesling et al ., 1988,1990年;冲向et al ., 1988,1990年)。堕落的寡核苷酸探针基于净化单元的结构被用来屏幕互补脱氧核糖核酸数据库,从而克隆α1β1-subunits。c端区域的子单元有一个高度的克隆序列的身份与腺苷和微粒guanylyl环化酶(包括),表明这是催化领域。硝普酸钠(SNP)敏感guanylyl环化酶活性对α1表示当克隆的互补和β1 cotransfected成不等的细胞系统,但不是当分别转染(Harteneck et al ., 1990;冲向et al ., 1990)。这些数据证明了国网公司的子单元所需的基底和nitrovasodilator-stimulated催化活性。
研究信号转导的包括提出了一个新的范式。海胆的精子是热解色谱的最大来源之一。在棘皮动物,肽分泌卵子的激活包括精子以特有的方式(铃木et al ., 1984;Ramarao和加伯,1985年)。此外,放射性标记的蛋肽可能是化学交联的精子细胞表面蛋白抗血清识别的大小一样,反对guanylyl环化酶(Shimomura et al ., 1986)。这些观察表明,热解色谱也可能作为肽受体配体。虽然这些研究被海胆,心房利钠肽(ANP)激活guanylyl环化酶,增加(cGMP)我在哺乳动物组织(原作者et al ., 1984;Waldman et al ., 1984;Winquist et al ., 1984)。随后,ANP绑定和guanylyl copurified环化酶活动,强烈建议两个活动驻留在单个分子(Kuno et al ., 1986;保罗et al ., 1987;Shimonaka et al ., 1987;Meloche et al ., 1988)。1988年,包括首次从海胆睾丸cDNA克隆库使用探测器基于胰蛋白酶的肽经纯化蛋白(辛格et al ., 1988)。这对隔离哺乳动物克隆提供了必要的探针互补编码包括。利钠肽受体,guanylyl酸环化酶(GC-A)和B (GC-B),是第一个克隆哺乳动物组织(包括Chang et al ., 1989;Chinkers et al ., 1989;劳et al ., 1989;舒尔茨et al ., 1989)。利钠肽受体的推断主要序列预测蛋白质用一个跨膜域划分一个细胞外配体结合域从胞内域。删除诱变研究已经证明,胞内域服务监管、二聚作用,催化功能(Chinkers和加伯,1989年)。这种监管域序列相似性与蛋白激酶,尤其是蛋白质酪氨酸激酶,单跨膜域受体(辛格et al ., 1988)。c端催化域的序列高度同源的α-和β-subunits国网公司和有限的身份两个催化域的腺苷酸环化酶(Krupinski et al ., 1989;索普和加伯,1989年)。
聚合酶链反应(PCR)的发展促进了搜索guanylyl酸环化酶家族的新成员。简并PCR引物基于保守氨基酸序列在国网公司的催化域和热解色谱用于优先放大guanylyl,与腺苷酸环化酶,产生了第二个α-序列和第二β-subunit国网公司和五个独特的包括序列(GC-C GC-G) (袁et al ., 1990;Harteneck et al ., 1991)。第三个对国网公司的子单元,克隆的筛选与鼠cDNA克隆人类cDNA图书馆,最有可能的人类直接同源α1 /β1 (Giuili et al ., 1992)。Guanylyl环化酶C (GC-C)的受体细菌热稳定肠毒素(STs) (舒尔茨et al ., 1990;德萨特et al ., 1991)和内源性肽guanylin和uroguanylin (Currie et al ., 1992;哈姆拉et al ., 1993)。剩下的克隆哺乳动物包括孤儿受体没有已知的细胞外配体。Guanylyl环化酶D (GC-D)表达在嗅觉神经上皮的带状模式类似seven-transmembrane域有气味的受体(Fulle et al ., 1995)。两个感官组织的其他成员的亚科guanylyl环化酶,guanylyl环化酶E (GC-E retGC-1)和guanylyl F环化酶(GC-F retGC-2),视网膜的表达(Shyjan et al ., 1992;劳et al ., 1995;杨et al ., 1995)。在松果体GC-E也表达了(杨et al ., 1995)。虽然这些酶是孤儿受体,细胞外的领域是同源的GC-D和分享类似的安排与其他包括半胱氨酸残基在细胞外的域。这表明他们可能有一个细胞外配体,尽管视网膜环化酶的催化活性是由Ca2 +]我通过guanylyl cyclase-activating蛋白质(GCAPs)。最近克隆GC-G最接近的利钠肽受体,尽管它不是激活钠尿肽(舒尔茨et al ., 1998 b)。显然,哺乳动物的家庭guanylyl环化酶是相对较小的,因为低紧缩库筛选和简并PCR在没有产生丰富的独特的互补。相比之下,秀丽隐杆线虫约有30种基因编码guanylyl cyclase-like序列和看似富含cGMP-coupled通路(Yu et al ., 1997)。
2。Guanylyl酸环化酶基因的结构和位置。
基因编码的染色体位点guanylyl酸环化酶的亚型及其配体被映射在人类和/或鼠标(表1,2)和分离,分散在整个基因组,也有明显的例外。因此,基因编码GC-A利钠肽配体,ANP和脑利钠肽(BNP)组织串联在人类和老鼠(黄et al ., 1996;田村et al ., 1996 b)。同样,guanylin uroguanylin,内生GC-C活化剂,通过密切相关基因进行编码(惠特克et al ., 1997)。视网膜guanylyl环化酶活动是由GCAPs,钙结合蛋白。到目前为止,三个联盟的成员GCAP家庭已确定。”GCAP1和发现GCAP2 tail-to-tail安排人力6号染色体,而GCAP3位于染色体3 (Subbaraya et al ., 1994;Haeseleer et al ., 1999)。
人类的基因编码国网公司子单元α3(相当于α1)和β3(相当于β1)映射到染色体4问(Giuili et al ., 1993)。因为子单元都需要在1:1化学计量学活动,他们共同的染色体位点可能意味着协调调节基因表达。基因编码α1和β1国网公司子青鳉鱼34-kb内同步组织(劳斯et al ., 1999)。的活动5′上游地区每个青鳉鱼的基因进行了分析使用绿色荧光蛋白记者在青鳉胚胎结构表达(劳斯et al ., 1999)。虽然α1上游地区推广的绿色荧光蛋白的表达,β1 5′地区不足,建议的表达α1β1-genes是协调一致的。然而,α2-subunit,也可以形成一个活跃的二聚体与β1体外,是由一种11号染色体上的基因编码(Yu et al ., 1996)。α2和β1-subunits二聚在生理条件下反对的协调监管要求表达α-和β-subunits (Russwurm et al ., 1998)。
几个基因的结构包括已确定,和他们的组织领域的保护体现在内含子/外显子安排。这样的安排是最高度保守的基因编码的部分催化和激酶同源域。的细胞外域guanylyl环化酶是守恒的,但不是之间,亚科和结构变化的部分基因。GC-A和- b基因相似的大小(16.5 - -17.5 kb)和结构,有22个外显子和几乎相同的内含子/外显子边界(山口et al ., 1990;Rehemudula et al ., 1999)。然而,内含子的大小并不是这些基因之间的守恒。同样,guanylyl环化酶在感官组织共享一个守恒的基因结构和只有20个外显子(杨et al ., 1996)。的基因GC-C要大得多(> 50 kb)比其他基因编码guanylyl环化酶和有一个独特的内含子/外显子安排(s .舒尔茨j .公园,s . a . Waldman未公开的数据)。国网公司亚基基因的结构尚未报道。
鲜为人知的监管guanylyl酸环化酶基因的表达。的基因的5′监管区域测序(GC-A, - c - e)没有典型的TATA盒和一个缺失或反向CAAT框。虽然许多通用转录因子结合位点存在共识,控制组织的元素表达现在才开始探索。GC-A基因启动子结合位点至少有三个共识Sp1,转录因子是涉及许多基因的表达在脉管系统(梁et al ., 1999)。化验使用情况和报告基因转移技术展示了这三个网站绑定Sp1和基底GC-A基因的转录是必不可少的(梁et al ., 1999)。表达的基因GC-A也受其配体,ANP。GC-A mRNA水平被抑制ANP在时间和浓度的方式培养主动脉平滑肌细胞(smc)和主要文化内髓集合管细胞(曹et al ., 1995,1998年)。cGMP cell-permeable模拟也抑制转录GC-A,建议第二信使,而不是利钠肽,负责调节基因活性(曹et al ., 1995,1998年)。启动子的ANP / cGMP-responsive元素GC-A尚未确定。
而GC-A表达在不同的细胞类型,在许多组织中,表达式的GC-C成年人似乎局限于肠道上皮和初级和转移性直肠癌(Carrithers et al ., 1996)。袋北美负鼠,guanylyl cyclase-coupled圣受体可能GC-C负鼠直接同源,表达在肾脏上皮细胞,肝、睾丸,气管和肠(福特et al ., 1989;伦敦et al ., 1999年版)。GC-C和绑定的mRNA圣中检测到放射性标记的新生儿和刚断奶的老鼠的肝脏,在胎儿、新生儿,再生鼠肝(兰妮et al ., 1992,1994年;Scheving和罗素,1996;斯文森et al ., 1996)。虽然敏感一技术已经被用于放大GC-C mRNA的许多组织,生产cGMP回应圣只有被观察到在啮齿动物的胃和内耳(肠外Krause et al ., 1997;伦敦et al ., 1997年版)。
的初始特征基因的5′侧翼区域GC-C,使用报告基因结构,建议intestine-specific转录活动在近128个基点(曼et al ., 1996 a)。这个区域的分析,这是守恒的人类和老鼠,发现潜在的几个转录因子结合位点。肝细胞的核4 (HNF-4)绑定到一个特定的元素GC-C近端启动子和刺激GC-C当转染细胞系的表达通常表示无论是GC-C还是HNF-4 (斯文森et al ., 1999)。HNF-4结合位点的突变废除GC-C的启动子活性肠细胞,证明HNF-4基底基因表达是必要的(斯文森et al ., 1999)。
最近的观察表明,转录因子Cdx2介导intestine-specific GC-C的表情。Cdx2属于homeodomain尾相关转录因子家族,果蝇需要蛋白质,其他几个基因的选择性表达在肠道组织(Traber西尔伯格,1996年)。删除或突变,近端GC-C Cdx2共识结合位点的基因启动子表达报告基因构造的活动能力降低在extraintestinal肠道细胞水平观察细胞(公园et al ., 2000)。
3所示。遗传病与Guanylyl环化酶。
唯一的人类疾病基因映射到guanylyl环化酶包括视网膜营养不良。雷伯氏先天性黑内障(LCA1),显性遗传性锥体杆体营养不良(CORD6),锥萎缩症(CORD5)和中央网状脉络膜萎缩症已经被映射到17号染色体p12-p13, GC-E区间包含基因(Balciuniene et al ., 1995;波瑞特et al ., 1996;休斯et al ., 1998;Kelsell et al ., 1998)。在LCA1 GC-E包含的基因突变,包括转移,这导致截断蛋白质缺乏kinase-like和催化领域由于过早终止翻译或一个错义突变kinase-like域(波瑞特et al ., 1996)。表达GC-E与这样一个错义突变异种的细胞系表明突变蛋白稳定但不激活GCAP1 (杜达et al ., 1999)。在CORD6 GC-E包含突变细胞内二聚作用域(Kelsell et al ., 1998)。这是假定这些突变可能会导致蛋白质空间变化,影响这两个突变体/变异和突变/野生型二聚体,从而导致CORD6的主要表型。事实上,其中一个突变体GCAP-1增加亲和力,产生一种酶,这种酶刺激(Ca更高2 +]我比野生型GC-E (塔克et al ., 1999)。因此,异常增加(cGMP)我在黑暗感光细胞可能会造成他们的退化。当老鼠的基因编码GC-E消除目标中断,锥消失了5周的年龄(杨et al ., 1999)。虽然棒的数量和形态从GC-E空老鼠类似野生型小鼠和暗电流是正常的,从零小鼠视网膜反应下降。矛盾的杆行为的原因尚不清楚。
基因改变guanylyl酸环化酶家族的其他成员不与任何描述人类的疾病表型。几个基因的编码guanylyl环化酶已经被目标功能消除老鼠破坏。这种方法可以提供洞察一个基因的正常生理作用的产品。GC-A基因的靶向破坏导致小鼠抗盐高血压(洛佩兹et al ., 1995;奥利弗et al ., 1997)。GC-A零老鼠无法注入ANP或者应对急性体积膨胀,刺激诱导利尿和尿钠排泄的野生型的同胞(岸本et al ., 1996)。空的老鼠心脏肥大。在一项研究中,所有GC-A零雄性老鼠死于充血性心力衰竭或主动脉夹层的6个月大的时候(奥利弗et al ., 1997;佛朗哥et al ., 1998)。因此,GC-A零老鼠表现出许多人类原发性高血压的特点和可能是一个有价值的模型来研究和开发治疗这种疾病。国网公司信号通路的变化也可能导致高血压的发展。在自发性高血压大鼠高血压动物模型,表达式α1——和β1-subunits国网公司和cGMP-dependent蛋白激酶的表达(PKG)我减少主动脉(Ruetten et al ., 1999)。减少表达观察即使在年轻的自发性高血压大鼠血压正常的人,暗示这是一个早期事件在疾病的发病机理。
该基因编码GC-C也遭受过破坏目标(曼et al ., 1997;舒尔茨et al ., 1997)。零老鼠可行和健康,抵抗感染产肠毒素的细菌,导致野生型小鼠腹泻和死亡。正常的生理作用GC-C因此仍是未定义的。
b膜结合Guanylyl环化酶
1。介绍。
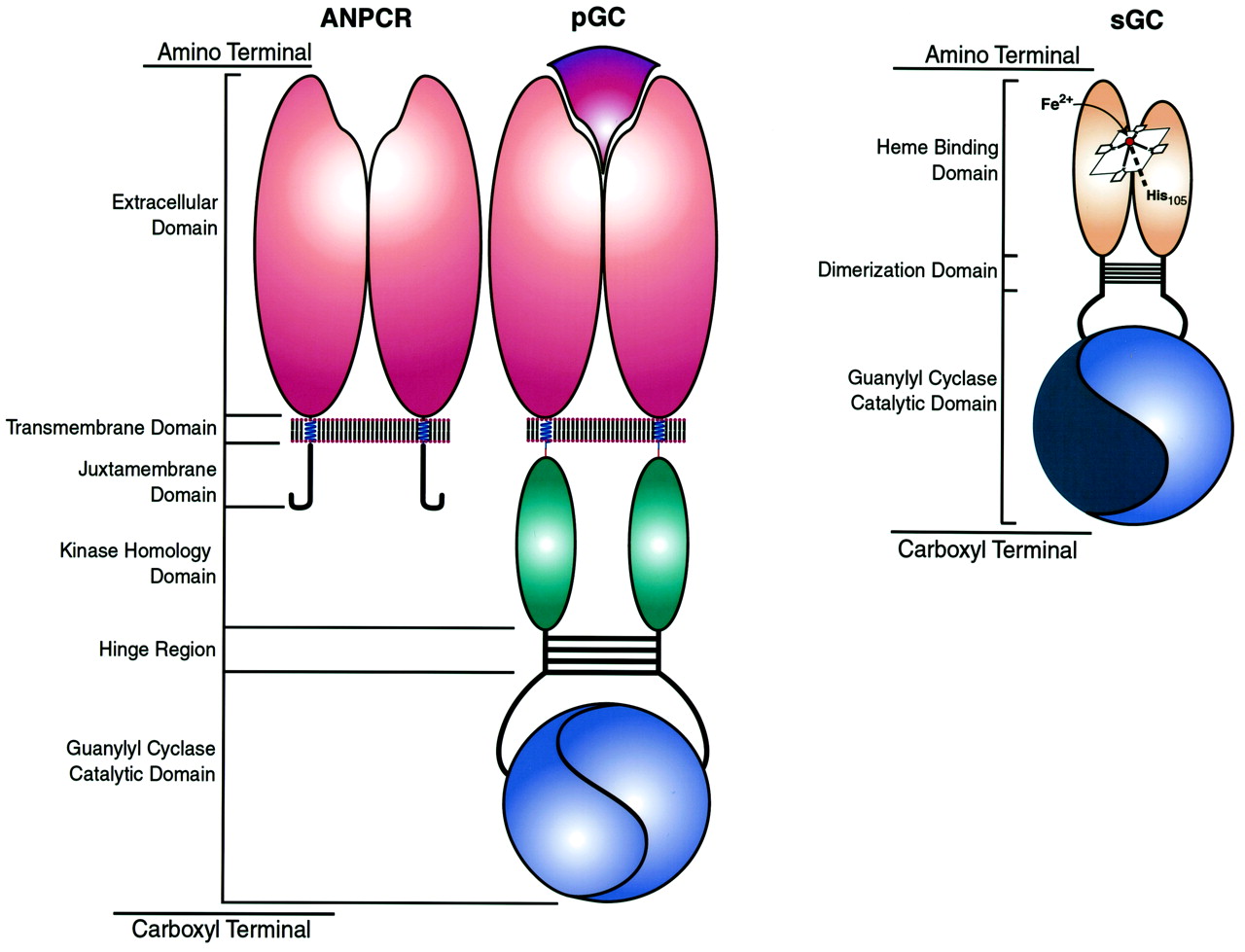
七真兽类哺乳动物包括亚型(GC-A GC-G)已确定(表2)。他们表现出高度保守的结构域,包括(1)一个细胞外绑定域定义的N末端,在某些情况下结合配体(GC-A, - b - c),(2)一个跨膜域,(3)细胞质近膜域中,(4)监管领域,显著的同源性蛋白激酶,铰链区(5),(6)c端催化域(无花果。1)。肠粘膜细胞亚型表达(GC-C)和感觉器官(GC-D, - e - f)也拥有c端尾巴。

域结构guanylyl环化酶。的同源域ANPCRs、热解色谱和国网公司进行了比较。ANPCRs是homodimeric截断guanylyl环化酶具有细胞外配体结合,跨膜,但缺乏激酶同源性和近膜域,铰链,催化领域。包括说明是为模仿GC-A - b和拥有一个由两个细胞外配体结合部位形成氨基末端域。除了域存在于ANPCRs,也包括具有激酶同源域,铰链区域,和催化领域,形成两个功能催化网站。GC-C - d, e, f具有羧基末端尾巴,这里没有描述。国网公司是拥有氨基末端形成监管域包含一个亚铁血红素辅基(Fe2 +)核心形成一个咪唑轴向债券与他105年β-subunit。此外,国网公司拥有二聚作用域和羧基末端催化领域,形成一个积极的和一个不活跃的(蓝色)催化部位。颜色识别领域有显著的同源性。
基于配体特异性,热解色谱分为(1)利钠肽受体,肠道peptide-binding受体(2),(3)孤儿受体(表2)。GC-A GC-B绑定和利钠肽被激活,包括ANP、BNP和c型利钠肽(CNP)。GC-C最初是作为家族的受体同源STs产生分泌性腹泻。最近,哺乳动物内源性肽,包括guanylin uroguanylin,和lymphoguanylin绑定并激活GC-C (Currie et al ., 1992;哈姆拉et al ., 1993;福特et al ., 1999)。GC-D - e, f, g是孤儿受体的配体仍有待确定。
包括表达在胎盘类哺乳动物(表几乎所有的组织2)。GC-A mRNA高度表达在肾、肾上腺、下级和脂肪组织,在回肠末端和人类胎盘(劳et al ., 1989)。GC-B mRNA丰富大脑、肺、肾(舒尔茨et al ., 1989)。GC-C已被确认的信使rna不仅在肠粘膜细胞在成年胎盘类哺乳动物,而且在许多上皮细胞在有袋动物(福特et al ., 1988;舒尔茨et al ., 1990;Carrithers et al ., 1996)。在中枢神经系统中,GC-D嗅感觉神经元表达族群,GC-E表达在视网膜视杆细胞和视锥细胞,而GC-F仅表现在视杆细胞(Fulle et al ., 1995;杨et al ., 1995)。GC-G主要是表现在肺,小肠,骨骼肌(舒尔茨et al ., 1998 b)。因此,利钠肽受体——环化酶,包括GC-A, b - g,是许多组织广泛表达。相比之下,GC-C和感官器官环化酶,GC-D, - e,和- f,所有拥有c端尾和组织的方式表达。
2。同形像的微粒Guanylyl环化酶。
利钠肽受体。
哺乳动物的心房心肌细胞产生ANP,介导的多效性的反应设计维护心血管体内平衡的压力或容量的挑战(大胆的,1985)。因此,ANP诱发尿钠排泄、利尿和低血压,抑制分泌肾素和醛固酮(de大胆et al ., 1981;Atarashi et al ., 1984 a,b)。事实上,ANP似乎调解短期和长期控制血压和液体和电解质平衡(大胆的,1985;约翰et al ., 1995,1996年;洛佩兹et al ., 1995;岸本et al ., 1996;奥利弗et al ., 1997;佛朗哥et al ., 1998)。除了ANP,其他钠尿肽鉴定日期包括法国巴黎和中国出版集团。ANP是合成prepro-polypeptide 151残留的c端部分包含了生物活性序列(大胆的,1985)。这种激素是成熟的循环形式28-amino酸组成的肽17-amino酸循环稳定由单个链内的二硫桥和N - c端扩展,都是生物活性所必需的。法国巴黎是一个26-amino酸肽,首次从酸提取分离的猪脑(Sudoh et al ., 1988)。随后,这种肽也被发现在心脏和血液(Aburaya et al ., 1989)。的disulfide-stabilized 17-amino酸环和c端序列,对ANP至关重要生物活性,在法国巴黎守恒。基因编码ANP和法国巴黎组织串联,在人类,ANP的法国巴黎位于上游的基因(黄et al ., 1996;田村et al ., 1996 b)。CNP 22-amino酸肽,首次发现在酸提取猪的大脑。CNP包含disulfide-stabilized 17-residue环结构中ANP和法国巴黎。相比之下,CNP缺乏一个c端扩展和氨基地区不是同源与ANP和法国巴黎。尽管CNP诱发尿钠排泄、利尿和血管平滑肌放松,它是更少的ANP、BNP (Sudoh et al ., 1990)。
利钠肽的主要信号机制引起的生理效应包括激活guanylyl cyclase-coupled受体和积累(cGMP)我。ANP对GC-A选择性亲和力,与GC-B相比,与half-maximal反应的浓度是前者比后者低1000倍(舒尔茨et al ., 1989)。ANP绑定GC-A已经证明了配体结合分析和关联交联研究Leitman et al ., 1988;Chinkers et al ., 1989;劳et al ., 1989;朱厄特et al ., 1993)。ANP (cGMP)增加我浓度和时间的方式在不同的细胞和组织(Chinkers et al ., 1989;舒尔茨et al ., 1989)。同样,ANP激活guanylyl环化酶在胞外膜准备从不同的细胞和组织(原作者et al ., 1984;Waldman et al ., 1984;Leitman et al ., 1987)。的浓度依赖性guanylyl环化酶激活和(cGMP)我积累引起ANP优于受体结合(黄et al ., 1995)。不等的细胞中表达GC-A缺乏内生发展的这种蛋白质的表达结果ANP专门绑定到这些细胞的能力,增加他们的包括活动,诱导细胞内积累(cGMP)我(Chinkers et al ., 1989)。ANP不绑定到组织动物GC-A零突变纯合子。这些动物不接受尿钠排泄或利尿ANP和主动脉环不放松,以应对肽(岸本et al ., 1996;洛佩兹et al ., 1997)。此外,ANP直接激活GC-A纯化从哺乳动物组织(Kuno et al ., 1986;Inagami et al ., 1991;Waldman et al ., 1991)。
内生为法国巴黎似乎也在GC-A受体,但法国的10倍低于ANP在刺激受体(Goeddel 1991)。巴黎银行增加guanylyl环化酶活性和积累(cGMP)我在细胞和组织模式,模拟ANP (Goeddel 1991)。同样,法国巴黎银行绑定到不同的表达GC-A并增加guanylyl环化酶活动和(cGMP)我积累在细胞(舒尔茨et al ., 1989)。主动脉瓣环从动物零突变纯合子的GC-A不放松对BNP (洛佩兹et al ., 1997)。
的主要配体GC-B CNP。尽管ANP和巴黎银行绑定到这个受体亲和力较低,CNP有50 - 500倍更大的亲和力GC-B比其他利钠肽(科勒et al ., 1991)。CNP刺激guanylyl环化酶活性和增加(cGMP)我在细胞和组织表达GC-B (森胁et al ., 1998;克里斯曼,加伯,1999年;道et al ., 1999)。同时,CNP结合细胞GC-B是不同的表达,增加活动的guanylyl环化酶和积累(cGMP)我在这些细胞(科勒et al ., 1991;克里斯曼et al ., 1993)。CNP诱发生理效应的动物GC-A零突变纯合子(洛佩兹et al ., 1997)。
b。肠肽受体Guanylyl环化酶。
GC-Coriginally被发现和克隆信使rna提取肠粘膜细胞(舒尔茨et al ., 1990)。尽管GC-C拥有guanylyl酸环化酶家族的保守域结构特征(图1),它不作为利钠肽受体。第一配体识别GC-C圣,圣GC-C激活和绑定guanylyl环化酶,增加(cGMP)我在肠道细胞(舒尔茨et al ., 1990)。圣是由细菌在肠道,包括大肠杆菌、肠杆菌属sp克雷伯氏菌。sp。,和鼠疫enterocolitica(Rao et al ., 1979;索恩et al ., 1979)。这个肽包含六个半胱氨酸形成三链内的二硫桥稳定的三级结构和授予:圣的特征(Guerrant et al ., 1980;Gariepy et al ., 1987)。还原二硫桥的消除了圣与受体结合的能力,促进跨膜信号传导,引起肠道分泌和腹泻(斯台普斯et al ., 1980)。GC-C似乎是唯一识别受体表达的圣在成人胎盘哺乳动物(Guerrant et al ., 1980)。圣结合GC-C浓度和时间的方式(Guarino et al ., 1987)。同样,GC-C激活圣浓度的方式,而不是受到利钠肽(舒尔茨et al ., 1990;Krause et al ., 1994 a)。特定的圣协会和GC-C已经证明了配体结合分析和关联交联(Guarino et al ., 1987;舒尔茨et al ., 1990;Hugues et al ., 1992)。交互的圣和GC-C激活guanylyl环化酶在胞外膜准备从肠道细胞(Gazzano et al ., 1991 a;Hugues et al ., 1991)。同样,圣诱发的积累(cGMP)我在细胞来源于肠(Guerrant et al ., 1980;Rao et al ., 1980)。不等的圣绑定表达式GC-C赋予特定的细胞(舒尔茨et al ., 1990;德萨特et al ., 1991;Deshmane et al ., 1995)。同样,圣激活guanylyl环化酶和诱发(cGMP)我积累在细胞异种的表达GC-C (舒尔茨et al ., 1990;德萨特et al ., 1991)。破坏的基因编码GC-C结果在小鼠抗ST-induced分泌性腹泻(曼et al ., 1997;舒尔茨et al ., 1997)。匀浆的肠膜GC-C零动物特别是绑定圣在非常低的水平,从而识别非GC-C结合蛋白(曼et al ., 1997)。然而,小说结合蛋白尚未确定或特征。因此,GC-C直接依然圣的唯一识别受体,介导的病理生理后果中的交互,包括分泌性腹泻。详细讨论postreceptor机制调解ST-induced肠道分泌在后面一节中介绍。
利钠肽受体不同,意义表达GC-C似乎局限于肠道粘膜细胞发现从十二指肠到直肠在成人胎盘哺乳动物(Krause et al ., 1994 a)。表达GC-C在肠道,但不是extraintestinal,细胞已经被圣绑定特定的刺激guanylyl环化酶,积累cGMP,检测特定的信使rna (Guerrant et al ., 1980;Guarino et al ., 1987;舒尔茨et al ., 1990;Hugues et al ., 1992;Krause et al ., 1994 a)。同样,肠粘膜细胞接受肿瘤转换后继续表达功能GC-C肠癌腺癌。检测这种蛋白质或其特定的信使rna extraintestinal网站似乎是一个敏感的和特定的标记检测转移性结直肠癌细胞(Carrithers et al ., 1996;Waldman et al ., 1998;Cagir et al ., 1999)。功能性GC-C也被发现在大鼠肝再生手术或化学肝切除术,但这个观察的功能意义仍有待阐明(兰妮et al ., 1994)。在胎盘类哺乳动物相比,GC-C有袋动物肠道上皮细胞的表达,胆囊、肺、肾、睾丸(福特et al ., 1988,1989年;白色et al ., 1989)。
ST-like肽结合和刺激GC-C隔绝哺乳动物。从肠道粘膜Guanylin和uroguanylin孤立;uroguanylin也已经从尿液分离(Currie et al ., 1992;哈姆拉et al ., 1993)。这些肽与圣分享显著的同源性,及其三级结构稳定的由两个链内的二硫键是至关重要的生物活性(Currie et al ., 1992;哈姆拉et al ., 1993)。精确的生理guanylin的重要性和uroguanylin还有待阐明。他们可能发挥作用在液体和电解质的调节肠道(哈姆拉et al ., 1993)。然而,老鼠零突变纯合子的GC-C似乎正常发育,正常的肠道功能(曼et al ., 1997;舒尔茨et al ., 1997)。Uroguanylin guanylin,圣似乎诱发尿钠排泄,利尿,kaliuresis啮齿动物肾脏。这些肽可能发挥作用在调节液体和电解质intestinal-renal轴(格林伯格et al ., 1997;Fonteles et al ., 1998)。然而,值得注意的是,敏感的嵌套一的分析没有检测肾脏GC-C mRNA的表达(Carrithers et al ., 1996)。最近,一种新型lymphoguanylin mRNA转录本编码,相关多肽guanylin uroguanylin,被发现在脾脏和淋巴组织从负鼠(福特et al ., 1999)。在这些研究中,cDNA lymphoguanylin是由孤立的RNA和用于合成一个假定的内源性肽。尽管圣、guanylin uroguanylin拥有多个二硫键,合成lymphoguanylin只包含一个链内的二硫键是生物活性(福特et al ., 1999)。因为这些研究没有隔离的内源性肽组织,这一发现的意义还有待建立。
c。孤儿受体Guanylyl环化酶。
热解色谱的内源性配体尚未确定归类为孤儿受体。配体激活其它guanylyl环化酶不激活GC-D, - e, f, g (Fulle et al ., 1995;杨et al ., 1995;舒尔茨et al ., 1998 b)。- e, GC-D的主要结构和- f细胞外的领域是同源,这些包括局限于感官的表达组织(杨et al ., 1995)。GC-G的配体可能像利钠肽,因为这包括同源的细胞外领域的利钠肽受体(舒尔茨et al ., 1998 b)。
3所示。结构的微粒Guanylyl环化酶。
细胞外的领域。
细胞外的领域包括展览至少家族成员之间的同源性。这种多样性在结构上可能反映了绑定的功能特异性和跨膜信号的感应不同的配体。的精确分子机制协调互动与细胞外配体激活的配体结合域和耦合催化域的定义。在GC-B, Glu332年似乎CNP绑定和所需的信号,因为它删除或替换他或赖氨酸结果完全环化酶活动的损失(杜达et al ., 1994)。然而,尚不清楚是否Glu322年对所需的受体蛋白质正确折叠至关重要配体绑定,或者如果它位于配体结合位点(杜达et al ., 1994)。同样,387到393在细胞外的域的氨基酸GC-C直接交互,需要绑定到圣(长谷川et al ., 1999)。
我,糖基化的受体。
所有哺乳动物包括,除了GC-F,包含至少一个N联系网站糖基化在细胞外的领域,尽管糖基化的程度不同受体(Chinkers et al ., 1989)。糖基化导致异质性的大小guanylyl酸环化酶受体。因此,脉冲追踪研究显示两个物种的GC-A合成在第7.5 h的孵化,但只有一个物种在孵化项目超过7.5 h。这两个物种中确定短孵化项目是混合糖化GC-A型,和更少的糖基化的受体作为完全糖化蛋白的前体(劳和保护,1992年)。同样,亲和标签与放射性标记的圣GC-C显示多个专门标记蛋白质,可以解决SDS-polyacrylamide凝胶电泳(页)(汤普森和Giannella, 1990;Hugues et al ., 1992)。也不等的表达GC-C产生多个受体蛋白特别标记与圣,展示他们来自一个成绩单翻译后加工(德萨特et al ., 1992;Vaandrager et al ., 1993 a,b)。大小不同的受体被转换为一个分子量通过与endoglycosidase治疗膜,删除N与碳水化合物(Vaandrager et al ., 1993 a)。同样,细胞生长在衣霉素,糖基化的抑制,生成一个受体物种(Vaandrager et al ., 1993 a)。
糖基化似乎扮演一个角色绑定包括细胞外配体的域。在进行的一项研究在人类胚胎肾细胞与GC-A稳定转染,完全glycoslyated GC-A专门ANP,但是碳水化合物残留的去除endoglycosidase阻止绑定ANP的受体(劳和保护,1992年)。在其他的研究在大鼠胶质瘤细胞与GC-A稳定转染,抑制N糖基化的GC-A衣霉素不改变ANP的能力与细胞结合并激活guanylyl环化酶(海姆et al ., 1996)。然而,衣霉素预处理抑制的反应GC-A ANP类似物,如urodilatin (ANP (95 - 126)) (海姆et al ., 1996)。不同的这两项研究的结果可能反映不同类型的ANP(本地或模拟)使用。同样的,当膜准备从细胞异种的表达GC-B endoglycosidase处理以去除碳水化合物蛋白质的糖基化的网站绑定CNP的膜迷路了(Fenrick et al ., 1996)。同时,活动的GC-B deglycosylated刺激条件下较低而充分的活动糖化受体(Fenrick et al ., 1996)。在关闭协议,deglycosylation endoglycosidase消除圣从细胞外GC-C域绑定(长谷川et al ., 1999 b)。每一个研究N与糖基化网站换成了Ala证明了Asn379年接近跨膜域,需要配体结合和催化活性长谷川et al ., 1999 a)。虽然供配体结合的糖基化是重要的,但它似乎没有要求分布的细胞表面受体(劳和保护,1992年;Fenrick et al ., 1996;长谷川et al ., 1999 b)。
GC-D包含两个潜在N与糖基化网站,而GC-E只包含一个(Fulle et al ., 1995;杨et al ., 1995)。GC-G包含五个潜在的细胞外的领域N与糖基化网站和大约是40%同源细胞外GC-A域(舒尔茨et al ., 1998 b)。是否这些孤儿受体的糖基化是必不可少的表达在细胞表面或转导信号尚未确定。
二世。半胱氨酸和受体齐聚。
所有哺乳动物包括有两个保守的半胱氨酸残基的氨基端和一个中途在细胞外的领域(Chinkers et al ., 1989;舒尔茨et al ., 1989,1990年,1998 b;Fulle et al ., 1995;杨et al ., 1995;促进et al ., 1999)。所有包括,除了GC-C,也包含两个保守的半胱氨酸残基在细胞外的糖基域近端membrane-spanning域(Chinkers et al ., 1989;舒尔茨et al ., 1989,1990年,1998 b;Fulle et al ., 1995;杨et al ., 1995)。GC-C股票两个半胱氨酸残基位于与GC-A中途在细胞外的域,但股票只有一个半胱氨酸残基与其他热解色谱(Chinkers et al ., 1989;舒尔茨et al ., 1989,1990年,1998 b;Fulle et al ., 1995;杨et al ., 1995)。从历史上看,有人推测,细胞外的半胱氨酸域形成链内的二硫键的重要稳定受体的三级结构,类似于其功能的其他成员总科的生长因子受体(Itakura et al ., 1994;Stults et al ., 1994)。ANP间隙受体(ANPCR)的GC-A截断同种型缺乏胞质域超出近膜域,拥有五个半胱氨酸在细胞外的域(劳et al ., 1990 a)(图。1)。特定场地诱变展示了前四个半胱氨酸顺序加入,形成半胱氨酸104年半胱氨酸132年和半胱氨酸209年半胱氨酸257年链内的二硫桥(Itakura et al ., 1994;Iwashina et al ., 1994)。这些的精确作用链内的二硫桥在细胞外的功能域和跨膜信号还有待定义。
半胱氨酸残基在细胞外域似乎调解ligand-independent齐聚反应的受体单体(Chinkers和威尔逊,1992年)。在ANPCR,第五个半胱氨酸在细胞外的域(半胱氨酸469年)是近端membrane-spanning域并形成一个稳定的跨链二硫桥二聚的结构(Itakura et al ., 1994)。不同的表达人类GC-A迁移时高分子量低聚物的复合物nonreducing条件下进行sds - page。这些低聚物的复合物可以转化为单体在sds - page减少条件下(劳,1992)。截短突变体的GC-A具有细胞外领域也形成低聚物的复合物,支持细胞外领域贡献的建议的半胱氨酸跨链二硫键调节寡聚化(劳,1992)。从牛肾上腺GC-A迁移作为一个四聚物的550 kda复杂当nonreducing条件下进行sds - page但140 kda单体当暴露于减少条件(岩田聪et al ., 1991)。GC-C迁移时高分子量复合物进行sds - page nonreducing条件下,但这些复合物被转换为单体接触减少条件(伊文思et al ., 1990)。也不等的表达GC-C形式转化为单体的高阶复合物接触减少条件(Vaandrager et al ., 1993 a,b)。
这些数据支持的建议,没有配体,包括自发形成稳定的复合物在细胞外二硫债券领域。那些有助于跨链的形成的半胱氨酸二硫桥仍然被定义为每个guanylyl酸环化酶的同种型。GC-A突变体,半胱氨酸423年附近membrane-spanning域所取代,自发形成的跨链二硫债券和接受二聚作用,大概由半胱氨酸432年未配对的突变蛋白(Labrecque et al ., 1999)。然而,这个观察本地受体的相关性尚不清楚,因为二聚在这个研究与本构活动(Labrecque et al ., 1999)。此外,GC-C也经历ligand-independent齐聚,并不拥有这些保守的半胱氨酸近端membrane-spanning域(长谷川et al ., 1999 b)。
b。跨膜域。
所有包括有一个跨膜域类似于生长因子受体超家族的其他成员。α-helix发现在跨膜域创建一个疏水区域,允许插入到疏水膜脂质双分子层。删除疏水性氨基酸的跨膜域表皮生长因子受体(EGFR)没有改变配体绑定或二聚作用,表明这一领域需要本地化的膜,但不是信号转导(Kashles et al ., 1988)。相比之下,删除的跨膜域转换生长factor-β受体改变其协调跨膜信号的能力(朱Sizeland, 1999)。除了膜插入,跨膜域可能促进通过α受体齐聚helix-helix交互(雷蒙和恩格尔曼,1994年;雷蒙et al ., 1994)。精确的角色的跨膜域包括外膜定位还有待定义。然而,值得注意的是,截短突变体GC-A GC-C,含有细胞外域但缺乏跨膜域,能够形成二聚体和结合配体(Chinkers和威尔逊,1992年;长谷川et al ., 1999 b)。
c。近膜域。
近膜域是一个短的地区约25个氨基酸远的胞质地区跨膜域蛋白质。虽然一个精确的函数没有被归因于这一领域,它可能涉及包括调解备用信号机制。这个地区包括包含一个共识序列存在于其他单跨膜域受体和重要的耦合heterotrimeric G蛋白(G蛋白)和下游效应器。
传统上,G蛋白受体的激活heptahelical家族(Gudermann et al ., 1995)。然而,单跨膜域生长因子受体超家族的成员,其中包括成员,也激活G蛋白和信号通过下游效应器。这些受体包括表皮生长因子受体、胰岛素样生长因子受体,胰岛素受体(Okamoto et al ., 1990;冈本和Nishimoto 1991;拉米雷斯et al ., 1995;Krieger-Brauer et al ., 1997)。所有的G protein-coupled单跨膜域受体序列在近膜域包含一个共识。这一共识序列与和激活G蛋白相互作用。共识序列的长度范围从14到20种氨基酸,包含两个基本残留氨基端和BBXXB主题c端端(B是一个基本的残渣和X是一个非基本,nonaromatic残留物)。GC-A,有趣的是,- b, c包含这一共识序列同源的位置在他们近膜域。
同样,ANPCR拥有上述共识序列在其近膜域(Fuller et al ., 1988;劳et al ., 1990 a)。这截guanylyl环化酶有一个短的37个氨基酸胞质域(劳et al ., 1990 a)(图。1)。ANPCR结合ANP、BNP和中国出版集团,它的主要功能似乎是间隙的利钠肽通过循环本构ligand-independent内吞作用(Nussenzveig et al ., 1990)。然而,这种受体也调节多种生理过程(Anand-Srivastava Trachte, 1993),包括抑制腺苷酸环化酶在大鼠和人类血小板(Anand-Srivastava et al ., 1991;马et al ., 1996),心房和心室注意到(Anand-Srivastava Cantin, 1986),大鼠心脏(Anand-Srivastava et al ., 1996),嗜铬细胞瘤细胞(Drewett et al ., 1992)。ANPCR也调节肝胚细胞瘤细胞(细胞增长·拉希德et al ., 1993)、扩散和入侵矩阵由内皮细胞(Pedram et al ., 1997),(Ca2 +]我在肾上腺球状带细胞(Isales et al ., 1992),激活内皮没有合酶在胃smc(以挪士)(没吃et al ., 1998),激活磷脂酶C-β3绦虫杆菌smc (没吃,Makhlouf 1999)和MAP激酶的抑制星形胶质细胞(普林斯et al ., 1996)。
的意义,近膜域需要ANPCR受体信号(没吃,Makhlouf 1999)。抗体针对ANPCR防止信号的胞质域域(Anand-Srivastava et al ., 1996)。此外,近膜域与Go在膜分离PC12细胞,可能调解抑制儿茶酚胺分泌ANP (Takida et al ., 1999)。同样,不同的表达ANPCR被耦合到激活coexpressed通过G以挪士我在胃smc (没吃et al ., 1998)。在头带杆菌smc、交互的ANPCR G我和激活磷脂酶的βγ-subunit C-β3 G我是由近膜域中共识序列。突变的序列消除间隙受体之间的耦合和激活磷脂酶C-β3 (没吃,Makhlouf 1999)。
这些研究表明,G protein-regulating近膜域中的上下文中共有序列一对guanylyl酸环化酶家族的成员可以与G蛋白和调节下游效应器ligand-dependent的方式。这一共识序列协调交替跨膜信号在其他单跨膜受体包括表皮生长因子受体、胰岛素样生长因子受体,胰岛素受体。这些观察表明,有趣的可能性,包括可能信号通过不同的途径,包括合成cGMP通过激活G的催化域和监管protein-coupled效应器通过共识序列近膜域。
d。激酶同源域。
我。结构。
之间的所有包括具有近膜和催化域残∼250 -激酶同源域(KHD),国网公司中是没有的。包括的KHDs∼30%同源与多种蛋白激酶(科勒et al ., 1992)。一般来说,蛋白激酶包含11守恒的子域和33不变的氨基酸激酶活性的关键(汉克斯et al ., 1988)。在利钠肽受体——环化酶、GC-A GC-B,和GC-G KHD拥有9日9日或8的守恒的子域和28日27日,分别或22不变的残留物。肠道受体环化酶GC-C包含8守恒的子域和25的33个不变的残留物,30%相对于GC-A守恒和GC-B (科勒et al ., 1992)。感官器官环化酶,GC-D, - e和f包含9日7或8的守恒的子域和22日21日或22日不变的残留物,分别。中一个不变的天冬氨酸残基子域名VI的蛋白质激酶,哪些功能是催化基础,取代了所有包括(Knighton et al ., 1991;泰勒et al ., 1992)。同样,glycine-rich地区的子域名我(GXGXXG)介导核苷酸结合蛋白激酶存在于GC-A - b,但在GC-C缺席。这种结构差异可能构成的一些功能性监管的差异这些受体由腺嘌呤核苷酸(科勒et al ., 1992)。
二世。激酶活性。
所有蛋白激酶在子域包含一个HRD共识序列VI的酸性Asp从ATP调节一个磷酸基的转移到适当的衬底(汉克斯et al ., 1988)。HRD的共识序列是缺席JH2域的木菠萝蛋白激酶家族,不表现出激酶活性(促进et al ., 1999)。同样,突变的不变的Asp Asn在c - kit和v-Fps导致亏损激酶活性(莫兰et al ., 1988;谭et al ., 1990)。这个催化Asp残渣代替热解色谱,一般到爵士,参数或Asn。因此,流行的假设是,包括不具有激酶活性,反映没有催化Asp残留的第六子域名(波特与亨特,1998 b)。尽管没有其他guanylyl环化酶具有蛋白激酶活性,视网膜包括展品内在激酶活性(Aparicio Applebury, 1996)。因此,ATP结合纯化视网膜guanylyl在网站不同于催化三磷酸鸟苷酸环化酶substrate-binding网站。蛋白激酶活性magnesium-dependent, autophosphorylates丝氨酸残基,并转移磷酸基髓磷脂碱性蛋白。的K米ATP是81μM,和环核苷酸激酶活动不受影响,佛波醇12-myristate 13-acetate (PMA),l-α-phosphatidylserine或Ca2 +,但被staurosporine抑制。这些属性是不同于其他对丝氨酸/苏氨酸激酶中确定杆外段包括蛋白激酶蛋白激酶C (PKC)和视紫红质激酶。这种观察内在的激酶和自身磷酸化活动包括仍然是一个孤立的结果,及其功能意义仍不清楚。
三世。磷酸化。
受体guanylyl环化酶作为磷蛋白质存在的基本状态。在绑定的配体,发生脱磷酸作用的受体,导致脱敏和减少ligand-induced guanylyl环化酶的活动。在KHD GC-A拥有六个磷酸化网站,包括爵士497年,用力推500年先生,502年先生,506年先生,510年,用力推513年,这是必要的GC-A ligand-dependent激活(波特和加伯,1992年;科勒et al ., 1993;波特与亨特,1998 b)。突变的这些网站单独Ala GC-A激活减少了ANP和替换的5个网站同时产生了一个对配体的受体。同样,GC-B作为磷蛋白质存在于基底状态时不绑定到受体配体。GC-B拥有五KHD内的残留物,包括爵士513年,用力推516年先生,518年先生,523年和爵士526年磷酸化的,(波特与亨特,1998 a)。GC-A, GC-B这些残留的磷酸化状态与催化反应和脱磷酸作用导致脱敏。阿拉巴马州这五个残基的突变导致CNP-dependent酸环化酶活性下降了90%,证明这些网站的磷酸化GC-B ligand-induced激活的是必要的。
e。铰链区。
铰链区43-residue域在GC-A KHD和催化领域。这个地区需要调节催化亚基的二聚作用以及酶活性的表达。截短突变体的GC-A只拥有催化域迁移在sds - page缺乏酶活性的单体(威尔逊和Chinkers, 1995年)。然而,截断突变体铰链区迁移的包容性为sds - page和拥有guanylyl环化酶催化活性(汤普森和加伯,1995年;威尔逊和Chinkers, 1995年)。铰链的一级结构域与卷曲螺旋配置一致支持特定蛋白质-蛋白质之间的关系(汤普森和加伯,1995年;威尔逊和Chinkers, 1995年)。的确,将这个序列整合到酵母2台混合动力系统展示了铰链域自发调节蛋白多聚体的形成(威尔逊和Chinkers, 1995年)。铰链区域介导催化亚基的二聚作用。二聚作用通常需要国网公司的催化活性,包括,腺苷酸环化酶(汤普森和加伯,1995年;威尔逊和Chinkers, 1995年)。此外,该地区可能会发挥更大的作用在调节holoreceptor二聚作用因为截断突变体GC-A缺乏胞质域未能形成二聚体或高阶结构在一项研究(劳,1992)。是否这个领域是必要的催化亚基的二聚或在调解过程中发挥作用受体齐聚在guanylyl环化酶除了GC-A还有待阐明。
f。催化领域。
我。二聚酶活性的催化领域是必需的。
Guanylyl环化酶表达催化活性必须经过二聚作用。Heterodimeric国网公司要求coexpression催化活性的α-和β-subunits和表达的亚基分别收益率催化地活性蛋白(Harteneck et al ., 1990;Buechler et al ., 1991)。截断的表达α和β催化域Sf9细胞产生催化地活动形成(威德尔et al ., 1995)。哺乳动物的腺苷酸环化酶催化域包含两个在单个多肽链(Krupinski et al ., 1989)。GC-A枢纽区,上面所讨论的,是绝对需要GC-A催化亚基二聚和表达催化活性(汤普森和加伯,1995年;威尔逊和Chinkers, 1995年)。同样,GC-C ligand-independent的方式形成低聚物,是重要的生产催化地活跃蛋白(长谷川et al ., 1999 b)。有趣的是,coexpressionα1-subunit国网公司和c端催化域的腺苷酸环化酶(I型、II或V),每个活动单独表达时,产生一个催化地活跃的腺苷酸环化酶(Weitmann et al ., 1999)。这种嵌合酶是由p区抑制剂,但不刺激Gα年代或forskolin。这些数据支持所需的建议两个催化域核苷酸环化酶的表达活动。同时,他们支持的建议guanylyl和腺苷酸环化酶的催化域结构上和功能上同源。
二世。嘌呤特异性的决定因素。
催化域的主要结构是高度保守的热解色谱和国网公司和催化领域密切相关的腺苷酸环化酶(Krupinski et al ., 1989)。因此,洞察的作用guanylyl环化酶催化领域得到使用的解决方案结构的x射线晶体鼠II型腺苷酸环化酶C2催化领域。这提供了重要的底物结合催化域信息(Tesmer et al ., 1997)。三个不变的残留物(赖氨酸、Asp、Gln)中腺苷酸环化酶的活性部位与嘌呤环相互作用,决定了底物特异性。老鼠国网公司的催化亚基(α1β1)包含三个不变的残留物(Glu、半胱氨酸、参数)职位同源的腺苷酸环化酶(图三个不变的残留物。2)。guanylyl和腺苷酸环化酶的突变,这三个残基被交换,交换了核苷酸底物特异性:guanylyl酸环化酶突变体特别是利用ATP作为衬底,而腺苷酸环化酶突变成为非选择性嘌呤核苷酸环化酶(Sunahara et al ., 1998)。这两个酶保留能力由特定的父核苷酸环化酶的活化剂。因此,营地生产的突变guanylyl环化酶是由国民党,而环核苷酸突变腺苷酸环化酶激活是由生产的Gα年代。同样,突变的视网膜guanylyl环化酶(Ret GC 1)基于腺苷酸环化酶的结构模型,Glu925年被替换下场赖氨酸和半胱氨酸吗995年Asp被替换下场,逆转从三磷酸鸟苷三磷酸腺苷(底物特异性塔克et al ., 1998)。这种变异保留特征能力的视网膜guanylyl环化酶是由GCAP-1 GCAP-2。

guanylyl酸环化酶活性的催化机理。三磷酸鸟苷结合到一个单一的催化部位在包括可溶性和两个催化网站。嘌呤环是一定会残留在β-subunit(红色),和二价阳离子代数余子式(我2 +),稳定的β-γ-phosphates核苷酸衬底,结合酸残留在α-subunit(绿色)。两个子单元贡献残留的催化中心协调乳沟α-phosphoanhydride债券通过单一的直接置换反应。催化是cGMP的产品和焦磷酸(PPi)。
三世。催化部位的配置。
腺苷的催化机理和模型guanylyl环化酶是晶体结构的基础上开发的老鼠II型腺苷酸环化酶C2催化域(刘et al ., 1997 b)(图。2)。该模型预测,heterodimeric环化酶如国网公司和腺苷酸环化酶有一个活性部位由两个催化亚基可以绑定一个底物分子二聚体(无花果。1,2)。Homodimeric环化酶,如包括有两个网站在一个裂口,每二聚体可以绑定两个底物分子(无花果。1)。三个残留需要形成一个催化中心,一个Asp从一个催化域和一个Asn /参数对。在heterodimeric环化酶,这两个领域形成一个催化中心,有一个领域提供Asp和其他领域提供Asn /参数对(无花果。2)。在homodimeric环化酶,每个域一个Asp和Asn /参数对形成两个催化中心。这个模型催化中心的形成与特征是一致的动力学行为guanylyl环化酶(Waldman和Murad, 1987)。纯化国网公司表现出线性米歇利斯情节希尔系数为1.0,与底物结合位点的一个类一致不互动(克里斯曼et al ., 1975;加伯,1979;Wolin et al ., 1982)。相比之下,净化包括展览曲线与希尔系数> 1.0米歇利斯情节,符合多个substrate-binding站点交互积极合作的方式(黄et al ., 1995)。
甘氨胆酸的,Glu974年代表一个不变的残留在所有克隆guanylyl环化酶和守恒的残留在大多数腺苷酸环化酶(威德尔et al ., 1997)。突变的残留在阿拉巴马州GC-A产生一种酶,这种酶是持续激活,对ANP和ATP。这些影响是KHD因为取代Glu独立的974年阿拉巴马州持续激活截断突变的GC-A缺乏细胞外领域,跨膜域和KHD。Glu的确切作用974年在热解色谱的调节机制还有待定义。
g。羧基末端尾巴。
GC-C和感觉器官guanylyl环化酶GC-D, - e, f包含c端尾,超越了催化域(舒尔茨et al ., 1990;Fulle et al ., 1995;杨et al ., 1995)。尾巴的精确功能还有待定义,尽管删除部分这一领域消除圣的能力来刺激生产GC-C cGMP的(和田et al ., 1996)。PMA强化激活GC-C圣,这是增加磷酸并入受体蛋白(Weikel et al ., 1990;起重机和小腿,1996;和田et al ., 1996)。然而,PMA没有加强圣激活或磷酸盐并入,截断突变缺乏c端22残留或替换Ser的突变1029年是阿拉巴马州所取代。这些观察表明,PMA诱导PKC-mediated磷酸化的Ser调节ST-dependent活动1029年在GC-C c端尾的。
有人猜测c端尾部可能参与将guanylyl酸环化酶受体与细胞骨架。GC-C, - e和f耐洗涤剂溶解,而GC-A和- b (别的Denisevich, 1979;别的et al ., 1980;Waldman et al ., 1986;Hakki et al ., 1993)。实际上,离液序列高的代理需要最佳溶解guanylyl环化酶膜制剂的肠上皮细胞(GC-C)或视网膜杆外段(GC-E GC-F) (别的et al ., 1980;Waldman et al ., 1986)。这个相对不溶性可能反映了协会的受体与细胞骨架环化酶,这可能是由c端尾巴,呈现他们耐火材料溶解洗涤剂。有趣的是,GC-C肠刷状缘膜和GC-E GC-F视杆外段局部膜专门化的共同起源(纤毛修改流程)由一个错综复杂的稳定和成熟的细胞骨架(1976年互联网)。
c端尾部可能调解的内化guanylyl酸环化酶受体。GC-C经历ST-dependent肠细胞的内吞作用(Urbanski et al ., 1995)。受体接受ligand-dependent受体介导内吞作用通常包含胞质域共识序列与内部设备交互。一个这样的共识序列YXXZ (Z是下面的疏水性氨基酸之一:L,我,V, M, C, A) (Johnson et al ., 1990;坎菲尔德et al ., 1991;托马斯和罗斯,1994年)。GC-C包含这一共识序列c端尾,这是假定调解ligand-dependent肠细胞的内吞作用(Urbanski et al ., 1995)。
4所示。Receptor-Effector耦合和微粒Guanylyl环化酶的功能。
中的交互的机制转化为催化活化和细胞信号和终止信号包括尚未完全阐明。然而,受体功能的细节,receptor-effector耦合效应激活,和信号终止如下所示。
配体和受体的相互作用。
平衡GC-C绑定特性的初步研究膜从肠道细胞表明圣将单个类的受体与摩尔Kd(Hugues et al ., 1991)。然而,进一步检查与广泛的圣浓度定义良好的平衡条件下显示两个种群的ST结合位点。受体展品的一个人口低容量(∼5%的受体)和高亲和力,而另一个受体展品人口高容量(> 95%的受体)和低亲和力(起重机et al ., 1992 b)。虽然圣高亲和力受体表现出强劲的配体结合,它们不是耦合guanylyl酸环化酶激活,及其功能意义仍有待定义(起重机et al ., 1992 b)。背后的分子机制的高和低亲和力受体仍不清楚,但可能反映受体齐聚,糖基化,或配体非均质性(德萨特et al ., 1992;Vaandrager et al ., 1993 a;舒尔茨et al ., 1998 a;长谷川et al ., 1999 b)。
圣低亲和力受体(GC-C)似乎同源受体功能耦合ANP (GC-A)。有趣的是,两个GC-A GC-C经过ligand-induced亲和力的转变,在跨膜信号似乎是重要的。因此,圣绑定诱发时间从高(0.1 nM)转移到低亲和力(1.0海里),和最低的亲和力GC-C状态似乎是受体亚型的功能耦合催化激活(起重机et al ., 1992 b)。同样,ANP绑定GC-A导致时间关联的转变。在平衡时,70%的ANP低亲和力受体存在状态(Kd= 2.5 nM),而30%留在高亲和力状态(Kd(= 0.3海里)然而et al ., 1991;朱厄特et al ., 1993)。
ligand-induced转变受体亲和力似乎KHD介导的。删除这一领域收益率GC-A, GC-C突变体“锁定”高亲和力的状态,无法接受转换到低亲和力状态(起重机et al ., 1992 b;朱厄特et al ., 1993;十行诗et al ., 1995)。ATP强化GC-A从高到低亲和力的转变引起的ANP绑定。增强作用的亲和力转变ATP被阿米洛利,竞争性抑制ATP结合蛋白激酶(海姆et al ., 1988;朱厄特et al ., 1993;十行诗et al ., 1995)。这种转变从高到低的亲和力与耦合中的交互guanylyl酸环化酶激活。GC-C锁在高亲和力状态由圣不激活,而入住率最低的亲和力状态是专门与ligand-sensitive激活(起重机et al ., 1992 a)。这些观察结果表明模型的配体结合的细胞外的领域包括诱导胞质域的变化,允许KHD ATP结合。腺嘌呤核苷酸与KHD去抑制催化领域,导致激活和随后的细胞外配体的亲和力下降域(波特与亨特,1998 b)。这个范例,其中中的交互激活下游效应器nucleotide-dependent方式与配体的亲和力下降,相关联特征,包括与G protein-coupled受体和效应器。在后一种系统,受体、效应和核苷酸监管职能驻留在单独的蛋白质,而在包括他们是单个域的函数在单个多肽。
b。齐聚反应的受体。
受体guanylyl环化酶表现出总体结构,类似于受体的酪氨酸激酶家族。酪氨酸激酶激活的接受模型包括一个要求ligand-induced二聚作用的受体单体。有趣的是,机制guanylyl环化酶激活偏离了这个模型。因此,包括似乎存在的寡聚物的基础状态,和中的交互不改变受体齐聚。预制受体寡聚物的要求没有配体可能反映出,在某种程度上,核苷酸的要求对两个催化亚基将核苷三磷酸腺苷酸环化酶循环核苷酸(Harteneck et al ., 1990;汤普森和加伯,1995年;燕et al ., 1996)。在热解色谱,GC-A - b, c, e存在低聚物(岩田聪et al ., 1991;Chinkers和威尔逊,1992年;十行诗et al ., 1995;长谷川et al ., 1999 b;Yu et al ., 1999)。
寡聚化GC-A一直是最广泛的热解色谱特征。共价交联凝胶过滤实验证明这两种单体的存在和高分子量复合物与受体寡聚物一致(Ishido et al ., 1986;岩田聪et al ., 1991;劳,1992)。GC-A免疫沉淀反应在缺乏ANP产生低聚物的复合物支持建议,这种受体self-associates ligand-independent时尚。结构缺乏激酶同源性和催化领域中单体基底状态,但在ANP的存在形成低聚物,这表明细胞质GC-A有助于寡聚化域(劳,1992)。然而,突变体缺乏细胞外域没有coimmunoprecipitate长篇GC-A,表明细胞外领域也是重要ligand-independent受体齐聚反应(Chinkers和威尔逊,1992年)。
尽管GC-C ligand-independent的方式形成低聚物,受体似乎接受ligand-dependent disulfide-stabilized二聚作用(Almenoff et al ., 1993;Vaandrager et al ., 1993 b,1994年)。受体中的标签和交联证明GC-C圣(存在于一个oligomerized国家独立Vaandrager et al ., 1993 b,1994年)。Coimmunoprecipitation不同的标记GC-C因为细胞表达证实寡聚化独立于配体刺激(Rudner et al ., 1995)。孵化与圣跨链导致GC-C二聚体的形成稳定的二硫键(Vaandrager et al ., 1993 b)。因此,GC-C可能存在于基底状态作为一个不活跃的homotrimer经历ligand-dependent内部重排形成催化地活动二硫交联二聚体(Vaandrager et al ., 1994)。有趣的是,GC-C突变体缺乏胞质域形成三只在配体的存在,与同源GC-A密切与结果突变体(劳,1992;长谷川et al ., 1999 b)。再一次,这些观察结果支持作用的胞质域调停ligand-independent包括寡聚化。
由腺嘌呤核苷酸c。监管。
我。别构激活Guanylyl环化酶核苷酸。
ATP强化激活GC-A和- b的利钠肽酶2 -三倍增加最大速度(V马克斯),而不改变底物亲和力(Kurose et al ., 1987;Chang et al ., 1990;Gazzano et al ., 1991 b)。欧共体50ATP增强作用的配体激活∼是0.1毫米,这是在生理范围内的细胞浓度的核苷酸(Kurose et al ., 1987;Chang et al ., 1990;Gazzano et al ., 1991 b)。核苷酸的等级次序的力量加强利钠肽受体激活腺苷′-O——(3-thiotriphosphate) (ATPγS) > ATP > adenylylimidodiphosphate (Chang et al ., 1990;Chinkers et al ., 1991;Gazzano et al ., 1991 b;福斯特和加伯,1998年)。的卓越疗效ATPγS和ATP可能反映了他们服务的能力既是:(1)变构的调节器guanylyl环化酶,和(2)基质蛋白激酶磷酸化调节的这些受体(福斯特和加伯,1998年)。相比之下,adenylylimidodiphosphate nonhydrolyzable模拟的ATP,这不是一个激酶衬底。增强作用的配体激活腺嘌呤核苷酸反映直接交互的核苷酸guanylyl环化酶,而不是nucleotide-dependent辅助蛋白质或酶。因此,除了nonhydrolyzable ATP的对应物,其他没有基质核苷酸或蛋白质的核苷酸激酶加强激活,包括ADP、腺苷′-O——(3-thiodiphosphate,腺苷′-O——(3-thiomonophosphate)(安培)(Kurose et al ., 1987;Chang et al ., 1990;Gazzano et al ., 1991 b;帕金森et al ., 1994)。同时,腺嘌呤核苷酸加强ANP激活GC-A纯化明显的同质性(然而et al ., 1991;黄et al ., 1995)。的确,GC-A杆状病毒表达或纯化同质性不能激活ANP在缺乏ATP,证明腺嘌呤核苷酸绝对需要receptor-effector耦合利钠肽受体(Chinkers et al ., 1991;然而et al ., 1991;黄et al ., 1995)。
除了对receptor-effector耦合的影响,腺嘌呤核苷酸也变构调节利钠肽受体配体的亲和力。GC-A接受ligand-induced时间从高(0.1 nM)转移到低亲和力(1海里)(Kd)(然而et al ., 1991;朱厄特et al ., 1993)。ATP的亲和力强这种转变与核苷酸对激活的影响。阿米洛利,拮抗剂ATP结合激酶催化领域,封锁了ATP对配体结合和催化活化的影响(朱厄特et al ., 1993;十行诗et al ., 1995)。事实上,阿米洛利“锁定”GC-A高亲和力对ANP的状态。这些数据表明,利钠肽受体的变构调节腺嘌呤核苷酸协调调节中的交互和receptor-effector耦合。
腺嘌呤核苷酸也加强GC-C的激活圣2 - 3重EC500.1毫米(Gazzano et al ., 1991 a;Vaandrager et al ., 1993 a)。效力等级次序的腺嘌呤核苷酸GC-C其余配体激活的类似的利钠肽受体,和上级效力ATPγS和ATP,与hydrolysis-resistant核苷酸相比,可能反映了他们的服务能力作为蛋白激酶变构调节器和基质。然而,在利钠肽受体相比,GC-C由圣绝对不需要ATP激活(Gazzano et al ., 1991 a;Vaandrager et al ., 1993 a)。同时,ATP不会改变V马克斯或底物亲和力GC-C (Vaandrager et al ., 1993 a)。相反,腺嘌呤核苷酸加强圣激活GC-C稳定酶的活性形式,防止其时间脱敏。GC-E和- f是类似于GC-C,他们不需要为催化ATP激活,但会使腺嘌呤核苷酸(塔克et al ., 1997)。像GC-C势差现象GC-E和- f的腺嘌呤核苷酸可能反映了保护催化失活(Vaandrager et al ., 1993 a;塔克et al ., 1997)。
变构调节催化热解色谱的激活和受体结合的腺嘌呤核苷酸似乎KHD介导的。突变体GC-A和GC-B缺乏KHD对腺嘌呤核苷酸和ANP (Chinkers et al ., 1991;科勒et al ., 1992;朱厄特et al ., 1993)。这些突变体的高亲和性的“冻结”状态,无法接受ligand-dependent转向低亲和力状态(朱厄特et al ., 1993)。实际上,这些突变体与野生型受体的结合特性阿米洛利处理(朱厄特et al ., 1993;十行诗et al ., 1995)。GC-A和- b拥有与共识序列GXGXXXG KHD glycine-rich的二级域名,介导核苷酸结合蛋白激酶催化领域中ATP的固定终端磷酸(汉克斯et al ., 1988)。突变体的GC-A和- b这个glycine-rich域是改变耐火材料配体的影响和腺嘌呤核苷酸(杜达et al ., 1993)。这些数据表明,变构调节GC-A和- b通过腺嘌呤核苷酸发生与KHD腺嘌呤核苷酸之间的相互作用,由中的交互和由KHD glycine-rich地区。Nucleotide-KHD交互传输信息远侧地的催化域和需要ligand-dependent催化活化。此外,信息传播向近端穿过膜效应转变为配体受体的亲和力域。有趣的是,GC-C缺乏glycine-rich KHD的子域名,不需要ATP配体激活的蛋白质,腺嘌呤核苷酸不改变催化的动力学,这些核苷酸中的不规范交互(Vaandrager et al ., 1993 b;Deshmane et al ., 1997)。值得注意的是,腺嘌呤核苷酸的作用guanylyl环化酶receptor-effector耦合类似于鸟嘌呤核苷酸的耦合heptahelical受体下游效应器。事实上,KHD热解色谱和G蛋白促进类似的功能出现在调停嘌呤核苷酸receptor-effector耦合在各自系统的监管。
二世。变构抑制Guanylyl环化酶核苷酸。
腺嘌呤核苷酸替换的两位嘌呤环,包括2-chloroATP和2-methylthioATP,抑制基底和ST-stimulated GC-C用K我(1)μM (帕金森et al ., 1994,1997年;帕金森和沃尔德曼,1996年)。抑制∼90%减少有关V马克斯,但它只有轻微影响酶对底物的亲和力。2-Substituted核苷酸并没有改变ST-induced cGMP积累在完整的肠细胞但permeabilized阻止这种效应细胞,这表明变构抑制不是purinergic受体介导的。核苷酸抑制由一个站点在分离和不同的受体,介导核苷酸激活(帕金森et al ., 1994)。有趣的是,guanylyl基体三磷酸鸟苷酸环化酶的效力增加2-substituted核苷酸抑制GC-C浓度的方式(帕金森和沃尔德曼,1996年)。此外,hydrolysis-resistant鸟嘌呤核苷酸模拟guanosine-5′-O——(3-thiotriphosphate)更有效的支持2-substituted核苷酸抑制GC-C相比之下,三磷酸鸟苷。此外,高浓度的三磷酸鸟苷模仿2-substituted核苷酸和直接抑制GC-C的影响。这些数据与模型一致的抑制GC-C 2-substituted核苷酸可能由一个附属鸟嘌呤核苷酸结合蛋白具有内在三磷酸鸟苷水解酶活动(帕金森和沃尔德曼,1996年)。虽然这个变构抑制内源性配体调控通路仍未定义,可以利用这种机制来阻止ST-induced液体和电解质分泌通过完整的肠细胞(Zhang et al ., 1999)。
d。激酶同源域。
KHD是一个关键的管理组件中的耦合互动效应包括激活。事实上,在这方面它的函数一样,类似于G蛋白质,这对夫妇heptahelical受体下游效应器。删除的KHD GC-A, - b和c导致本构激活的酶(Chinkers和加伯,1989年;科勒et al ., 1992;Rudner et al ., 1995)。同时,突变体受体缺乏KHD被腺嘌呤核苷酸和配体对刺激。此外,GC-A和- b缺乏KHD高亲和性“锁定”状态,对腺嘌呤核苷酸的影响受体的亲和力,和无法接受ligand-dependent转向低亲和力受体的特征(朱厄特et al ., 1993)。这些数据符合的模型KHD行为的抑制因子催化域的基底状态(Chinkers和加伯,1989年;科勒et al ., 1992;朱厄特et al ., 1993;Rudner et al ., 1995)。中的相互作用导致的变更KHD允许腺嘌呤核苷酸约束力,除抑制催化域反射激活,减少配体的受体的亲和力。值得注意的是,KHDs似乎从个人亚型包括功能具体为每个同种型。因此,KHDs GC-A - b,结构具有相似功能的同源蛋白质,可以在不改变这些受体的能力交换对腺嘌呤核苷酸和利钠肽(科勒et al ., 1992)。然而,交换的GC-A KHD GC-C或EGFR激酶结构域的产生酶,对利钠肽(科勒et al ., 1992)。这些数据表明,KHDs结构和/或功能“匹配”具体包括亚型。这个组件的兼容性的潜在分子机制还有待阐明。
e。磷酸化和同源和不同的脱敏。
早期研究表明,海胆精子包括接受与同源交互激活快速脱敏紧随其后在蛋肽(加伯,1989)。这些受体在基底高度磷酸化状态,和中的交互导致大规模的去磷酸化。激活这些受体的配体要求他们完全磷酸化。因此,治疗这些准备工作与磷酸酶产生对配体的受体。此外,脱磷酸作用中的相关受体相互作用导致受体的脱敏。同样,GC-A和- b要求ligand-dependent催化活化受体磷酸化(波特和加伯,1992年,1994年;科勒et al ., 1993)。纯化酶制剂的GC-A磷酸化保留完整的敏感性利钠肽和腺嘌呤核苷酸(福斯特和加伯,1998年)。此外,磷酸酶GC-A导致脱敏治疗和一个配体无法激活催化域(波特和加伯,1992年)。这些数据表明,磷酸化调节热解色谱是一个重要的机制。在基底状态,包括磷酸化,ligand-induced需要激活。中的互动与催化相关受体的激活启动一连串导致脱磷酸作用导致同源脱敏。有趣的是,这种机制的同源脱敏是反过来调节heptahelical G protein-coupled受体,磷酸化在基底状态但麻木的kinase-mediated磷酸化,诱导中的作用(Gudermann et al ., 1995)。
磷酸化网站重要的配体激活和同源脱敏已确定的KHD GC-A和- b (杜达et al ., 1993;波特与亨特,1998 a,b)。然而,激酶(s)负责维护GC-A和- b在基底的磷酸化状态尚未确定。同样,磷酸酶(s)负责ligand-induced GC-A和脱磷酸化- b调停同源脱敏尚未确定,尽管证据guanylyl cyclase-associated磷酸酶提出之前(Chinkers 1994)。虽然这种机制似乎可概括的海胆精子热解色谱和利钠肽受体环化酶,同源脱敏脱磷酸作用没有被证明的其他包括亚型。
GC-A - b也接受由数量不等的脱敏激活PKC的配体,包括PMA、内皮素、血管加压素和血管紧张素(波特和加伯,1994年)。激活PKC解开GC-A和- b ligand-induced cGMP生产(贾斯瓦尔et al ., 1988;波特和加伯,1994年)。但是,与其他受体系统,GC-A和- b似乎麻木了PKC-mediated脱磷酸作用(波特和加伯,1994年)。的确,PMA、直接激活PKC诱导相关的去磷酸化的利钠肽受体受体脱敏(波特和加伯,1994年)。特定的PKC抑制剂阻止脱磷酸作用和脱敏PMA诱导的ANP (波特和加伯,1994年)。此外,PMA治疗导致脱磷酸作用的受体残留不重叠与ANP残留脱去磷酸。这些数据表明GC-A和- b进行异种的脱敏脱磷酸作用介导的残留不同于那些参与同源脱敏(波特和加伯,1994年)。精确的机制不同的脱敏和PKC是否刺激导致包括激酶的抑制或激活的热解色谱磷酸酶仍有待定义。
f。辅助蛋白的监管。
视网膜热解色谱(GC-E - f)调制的家庭calcium-regulated辅助蛋白质,GCAPs。在视觉上过程中,光phototransduction成电脉冲发生在视网膜(Lolley和李,1990年)。包括位于外段级联中视网膜膜中央调停phototransduction (Lolley和李,1990年)。激活视紫红质耦合到一个G蛋白激活cGMP-specific PDE (Yarfitz赫尔利,1994年)。这PDE水解cGMP,导致关闭cGMP-gated渠道,调解Ca2 +涌入。减少(Ca2 +]我联盟协调GCAP激活GC-E”和- f和cGMP的生产(Shyjan et al ., 1992;劳et al ., 1995;Nakatani et al ., 1995)。因此而积累的cGMP重开cGMP-gated通道重建静止状态(黑暗)电流。
GCAPs份额显著的同源性与钙调蛋白家族的钙结合蛋白(Palczewski et al ., 1994)。到目前为止,三个哺乳动物GCAPs已确定(Palczewski et al ., 1994;Dizhoor et al ., 1995;Gorczyca et al ., 1995;Haeseleer et al ., 1999)。这些GCAPs展览guanylyl环化酶特异性:GCAP-1只激活GC-E,而GCAP-2激活GC-E和GC-F (Gorczyca et al ., 1995;Otto-Bruc et al ., 1997 a,b)。一般来说,GCAPs激活guanylyl环化酶(Ca2 +]我< 300 nM和抑制活动时(Ca吗2 +]我> 500海里(Dizhoor赫尔利,1996年;Dizhoor et al ., 1998)。尽管GCAPs调节guanylyl calcium-dependent地环化酶活动,协会的GCAPs guanylyl calcium-independent环化酶(Dizhoor赫尔利,1996年;劳拉和赫尔利,1998年)。
GCAP激活视网膜guanylyl环化酶已经被两个机制:建议发生二聚作用的增强视网膜guanylyl GCAPs通过二聚环化酶催化亚基,和/或GCAP-mediated稳定三磷酸鸟苷绑定到催化亚基(Olshevskaya et al ., 1999;索et al ., 1999)。二聚模型表明:(1)GCAPs绑定到视网膜guanylyl环化酶(Ca的独立2 +]我,(2)在高(Ca2 +]我,GCAPs绑定2 +和self-association亲和力较低,(3)在低(Ca2 +]我从Ca, GCAPs分离2 +联盟和结伴,(4)GCAP homodimerization”促进热解色谱二聚作用,因此提高催化活性(Olshevskaya et al ., 1999)。这个拟议的机制提供了一个合理的解释guanylyl calcium-sensitive监管的GCAPs环化酶,但是进一步的工作定义联盟协会GCAP和guanylyl”环化酶二聚作用是必需的。
虽然已确定多个联盟潜在GCAP结合位点”,到目前为止只有一个结合位点的视网膜guanylyl环化酶催化域特征(索et al ., 1999)。有趣的是,这种结合位点相同地区的腺苷酸环化酶催化域激活介导的Gsα(Tesmer et al ., 1997;索et al ., 1999)。Gsα激活腺苷酸环化酶催化部位的稳定过渡态与ATP (Tesmer et al ., 1997)。同样,有人建议GCAPs可能调节催化活性稳定guanylyl cyclase-GTP过渡状态(索et al ., 1999)。之间虽然有很大的同源性视网膜的结构和其他包括辅助蛋白质调节非视网膜guanylyl环化酶仍有待确定。
g。耦合模型颗粒Guanylyl酸环化酶受体和效应。
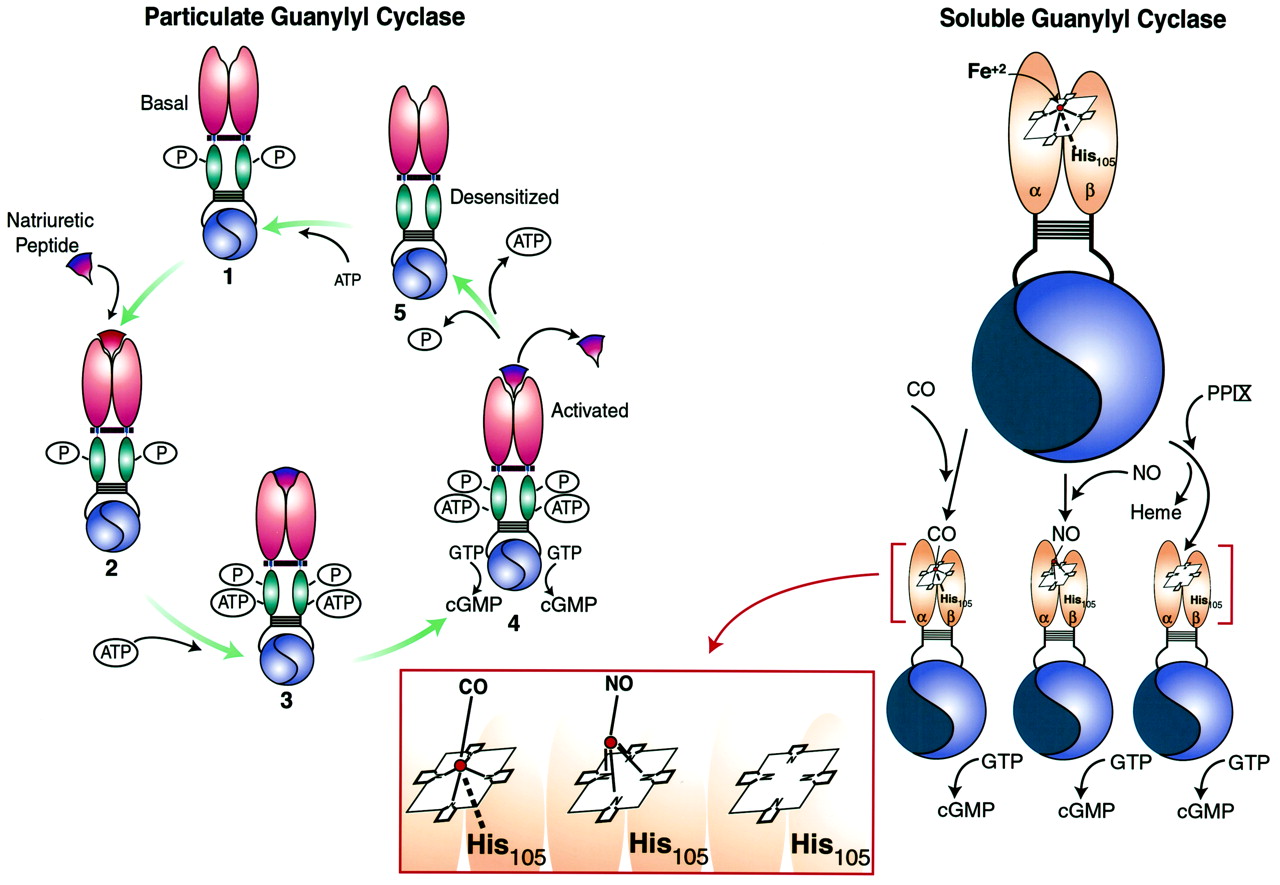
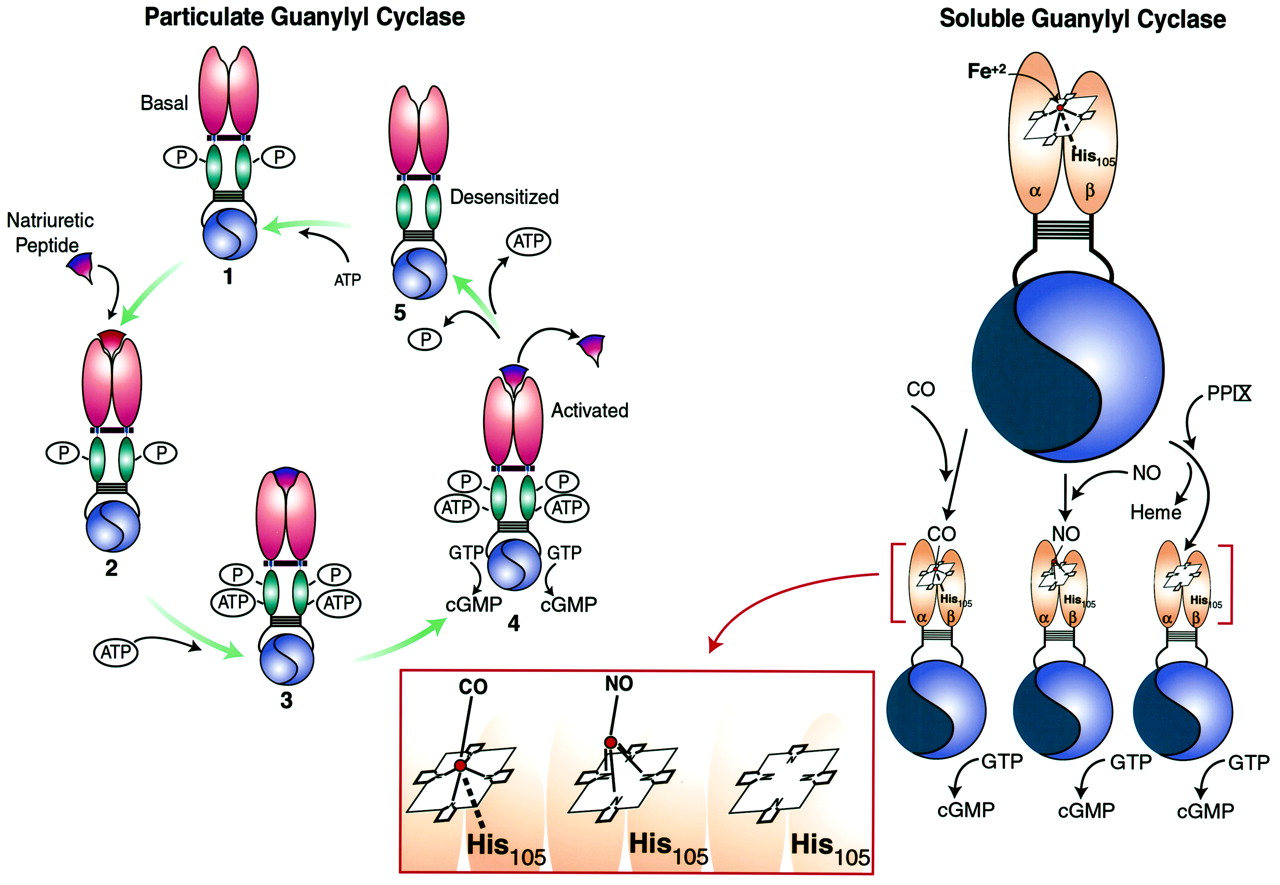
上述讨论表明以下receptor-effector耦合的一般模型为配体的监管包括(无花果。3)。在非活动状态,包括存在作为self-associated homo-oligomerized复合体没有配体。复杂中的每个单体在关键的丝氨酸或苏氨酸残基磷酸化glycine-rich周围的区域在KHD ATP结合位点。齐聚反应和磷酸化细胞外绑定域名要求的高亲和力的状态和存在于胞质域主管应对中的作用。在这些条件下,高亲和力配体结合网站是通过质膜的定义机制转化为一个KHD变更,导致ATP的绑定到域。协会的ATP KHD启动三个重要过程。首先,KHD压制的催化热解色谱领域基础状态,和腺嘌呤核苷酸与KHD交互导致脱抑制,反映了激活的催化域和增加V马克斯cGMP的生产。除抑制催化域的可能与内部重组有关的homo-oligomers跨链二硫键的形成,而不是整体破坏的寡聚物单体。第二,ATP与KHD协会是通过等离子体膜译成减少细胞外绑定域名配体的亲和力。第三,有时间去磷酸化KHD导致腺嘌呤核苷酸同源受体的脱敏和配体。中的精确触发启动去磷酸化,包括互动,腺嘌呤核苷酸KHD绑定,和/或激活催化域的尚不清楚。终止信号反映了三个过程,包括从细胞外配体分离领域,减少细胞外的亲和力域由腺嘌呤核苷酸KHD绑定,和同源受体的脱敏KHD脱磷酸作用。在这个模型中,要求receptor-effector耦合包括受体齐聚,KHD磷酸化,配体的入住率细胞外绑定域名,KHD和腺嘌呤核苷酸绑定。未能满足这些需求导致解偶联受体和效应器。尽管上述过程提供作为热解色谱receptor-effector耦合的一般模型,需要注意的是,它是专门研究产生的利钠肽受体。的确,GC-C并不拥有KHD glycine-rich ATP绑定域名,不需要ATP GC-C receptor-effector耦合,而且没有明显的角色KHD脱磷酸作用和同源受体脱敏。 In addition, the other known pGCs are orphan receptors, with no defined ligands. Although the above process is specifically referable to natriuretic peptide receptors, it serves as a working model to study the other members of the family.

调节微粒和可溶性guanylyl环化酶配体。微粒guanylyl环化酶:配体调控的模型包括描述是基于监管GC-A的利钠肽。在基底unliganded状态,包括存在self-associated homo-oligomerized复杂的(1)。复杂中的每个单体在关键的丝氨酸或苏氨酸残基磷酸化在KHD之内。齐聚反应和磷酸化细胞外绑定域名要求的高亲和力的状态和存在于胞质域主管应对中的作用。在这些条件下,利钠肽绑定到一个高亲和力的网站由为通过质膜(2)被翻译成一个KHD变更,导致ATP的绑定域(3),在基底状态,KHD压制催化热解色谱领域,和ATP交互领域导致脱抑制,反映了两个功能的激活催化域和增加V马克斯cGMP生产(4)。然而,协会的ATP KHD也降低了亲和力的细胞外绑定域名利钠肽和诱发KHD脱磷酸作用,导致ATP和配体的解离,导致脱敏(5)。再生反应的受体在基底状态配体可能需要rephosphorylation KHD的网站。可溶性guanylyl环化酶:国网公司,形成具备pentacoordinated (Fe亚铁血红素辅基2 +)核心与轴向配体咪唑在他有关105年β-subunit。没有激活国网公司通过直接绑定到第六的位置亚铁核心,打破债券之间的铁和组氨酸,游离于轴向咪唑配体,取代铁卟啉环的从飞机上。这将创建一个结构类似PPIX,国网公司的强有力的催化剂。终止离解的激活可能结果没有轴向配位位置的铁芯的血红素和返回的飞机卟啉环。公司结合国网公司的血红素组,产生一个hexacoordinated复杂的铁芯与咪唑和轴向配体同时进行。公司无力分离轴向配体咪唑形成pentacoordinated复杂不允许位移的血红素铁卟啉环的平面。因此,公司是国网公司更有效的作为活化剂,相比之下,没有。PPIX激活国网公司的确切机制仍不清楚。在一个模型(描述),PPIX取代国网公司的血红素,导致激活。另外,sGC-bound血红素铁可能交换2 +与自由PPIX,导致自由血红素,入住率PPIX监管领域的酶,和激活。
c .可溶性Guanylyl环化酶
国网公司几乎所有哺乳动物细胞的细胞质中的表达和介导多种重要的生理功能,如抑制血小板聚集,平滑肌松弛,血管舒张,神经元信号转导、免疫调节(科利尔瓦兰斯,1989)。这种酶是一种heterodimeric蛋白组成的α-和β-subunits和表达需要两个亚基的催化活性(Kamisaki et al ., 1986;Harteneck et al ., 1990;Buechler et al ., 1991)。每个单元都有一个氨基端管理域和c端催化域与相应的域共享序列同源性微粒guanylyl和腺苷酸环化酶(Chinkers et al ., 1989;Krupinski et al ., 1989;索普和Morkin, 1990)。
1。可溶性Guanylyl酸环化酶的亚基结构和同形像。
分析不同组织了多个同形像的国网公司有不同的亚基组成。最丰富的子单元α1β1,发现在许多组织(Braughler et al ., 1979;加伯,1979;Lewicki et al ., 1980)。这些亚基是第一只克隆鼠和牛肺(Koesling et al ., 1988,1990年;冲向et al ., 1988,1990年)。分别表达α1和β1收益率不具有催化活性的蛋白质,而coexpression这些单元的收益率国网公司,它可以激活没有(Buechler et al ., 1991)。β2-subunit,克隆鼠肾脏,包含86个额外的氨基酸与β1相比,c端地区(袁et al ., 1990)。这些额外的序列包含一个共识序列(CVVL)参与转录后修饰,如isoprenylation carboxymethylation,表明β2可能本地化国网公司膜。这∼76 kda亚基在肾脏和肝脏是最丰富的。虽然β2-subunit可以用α1形成异质二聚体,这个全酶表现出较低的比活度与α1 /β1。因此,无刺激COS-7细胞cotransfectedα1 /β1导致三倍cGMP生产比细胞转染α1 /β2 (古普塔et al ., 1997)。Coexpression的β2α1β1减少α1的形成/β1异质二聚体,可能由于β1之间的竞争和β2α1绑定。这些数据支持了假设的表达β2可以调节α1 /β1国网公司活动。事实上,表达β2已建议在高血压的发病机制中发挥作用在达尔鼠(古普塔et al ., 1997)。∼82 kdaα2克隆人类胎儿大脑与β1形成或β2,但降低β1的亲和力。事实上,α2 /β1比活度低于α1 /β1 (Harteneck et al ., 1991)。另外两个人类国网公司的子单元,α3 (82 kDa)和β3 (70 kDa),从成人的大脑已经被克隆。尽管α3和β3同源的氨基端区域有限,他们之间有72%的同源性残310 - c端区域(Giuili et al ., 1992)。同源区域的存在可以反映出一个共同的祖先的存在对这些单元(Giuili et al ., 1992)。
RNA拼接也促成了国网公司的子单元的异质性。的一种变体α2-subunitα2我,被发现在许多细胞系和组织使用基于保守序列的引物PCR在哺乳动物guanylyl酸环化酶的催化域(Behrends et al ., 1995)。子单元α2我是通过可变剪接的RNA,增加了31个氨基酸催化领域,与同源性催化领域内的一个区域腺苷酸环化酶。先前的研究证明了国网公司催化ATP营地的转换(米塔尔的Murad, 1977)。该地区α2的同源性我假设增加的能力这个同种型国网公司利用ATP的衬底和生产营地(Behrends et al ., 1995)。然而,coexpressionα2我/β1 Sf9细胞废除国网公司的能力产生营(Behrends et al ., 1995)。此外,与α2 Sf9细胞转染我/β1缺乏guanylyl环化酶活性,而coexpression cGMPα2 /β1导致生产的这些细胞,表明α2我亚基可以与α2争夺绑定β1和作为一个占主导地位的消极的抑制剂。这个亚基的表达可以作为一种机制来调节特定细胞(国网公司活动Behrends et al ., 1995)。此外,两种形式的人类国网公司的β-subunit, HSGC-1和HSGC-2肺中发现使用PCR和寡核苷酸引物从鼠和牛国网公司相应的保守序列(Chhajlani et al ., 1991)。HSGC-1是相同的与小亚基在牛和鼠肺,而HSGC-2包含33-amino酸删除。国网公司的剪接变体可能反映组织酶的具体表达式,以及活动的调节机制。
国网公司的小说对碘氧基苯甲醚,不需要异质二聚体的形成来表达催化活性被孤立的烟草天蛾的幼虫蛾的神经系统Manduca sexta。这部小说同种型、MsGC-β3老鼠β1-subunit密切相关(Nighorn et al ., 1999)。然而,MsGC-β3包含一个额外的315个氨基酸糖基,不与任何已知的同源蛋白质。MsGC-β3缺乏那些保守序列存在于其他国网公司重要酶的激活。MsGC-β3 COS-7表达细胞的催化活性小于MsGC-α1和MsGC-β1亚型。MsGC-β3活性弱的SNP,无法产生。然而,就像所有其他已知的环化酶,MsGC-β3 Mn时表现出较高的酶活性2 +,而不是毫克2 +,用作金属衬底代数余子式(Nighorn et al ., 1999)。
除了MsGC-β3,第二个新的guanylyl环化酶被隔绝的可溶性部分细胞的神经系统Manduca sexta。MsGC-I类似于热解色谱,GC-B最大的同源性。然而,它并不包含一个信号序列或配体结合,跨膜和激酶同源域包括哺乳动物的特征。尽管这种酶的位置是假定在细胞的细胞质,MsGC-I没有相似的监管领域的α-和β-subunits国网公司。它包含一个149个氨基酸序列扩展催化域之外,没有任何已知的同源蛋白质。MsGC-I因为细胞中表达水平较高的形式为基底guanylyl环化酶活动,但不能激活SNP。这样一个事实:一个细胞外配体或没有不能激活这种酶显示新的监管机制的存在这guanylyl环化酶(辛普森et al ., 1999)。
虽然天然为没有孤立的,广泛的sGCα之间的同源性和sGCβ子单元表明可能性(威尔逊和Chinkers, 1995年)。谷胱甘肽S-transferase(销售税)标记的重组人类α1——和β1-subunits可以形成homodimeric GST-α1 /α1和GST-β1 /β1 Sf9昆虫细胞复合物。Cotransfection补充子单元的形成导致催化地活跃,虽然是为检测的。为都是催化地活动,发现在数量低于形成。异质二聚体形成的偏好Sf9细胞可能反映了更高的亲和力之间互补的子单元。这些结果表明生理平衡人类的可能性,形成能够调节细胞(国网公司活动查贝尔et al ., 1999)。虽然两个互补的子单元的存在是必要的对于催化活性,这些亚基可能不是表达了相同的时间模式。在老鼠大脑,α1表达比β1早在胎儿的大脑发育。这个观察表明存在但一个个β-subunitβ1替代品在这个发展时期(Smigrodzki和莱维特,1996)。
2。域结构。
国网公司的每个单元可分为三个功能域:heme-binding、二聚作用,催化(无花果。1)。heme-binding域位于N每个亚基的终点站。血红素辅基的存在需要激活的国网公司没有(懦夫和DeRubertis, 1978,1983年;Gerzer et al ., 1982;Ignarro et al ., 1982 a;Ohlstein et al ., 1982)。血红素是一个五元环氮含量在四个氮原子与一个中心协调铁,可以是铁的2 +(或减少亚铁形式)或铁3 +(铁或氧化形式)(图1)。第五个成员国网公司是一个咪唑环的轴向配位体协调β1-subunit在他105年(石头和Marletta, 1994年)。突变的组氨酸,位于Nβ1-subunit的终点站附近,结果在国网公司无力血红素结合并产生一种酶,这种酶是没有响应。突变的守恒的组氨酸并不影响酶的能力结合血红素(威德尔et al ., 1994)。这种酶突变在他105年与血红素可以净化和重组,但它仍然是对不。
α1——和β1-subunits都需要表达基底国网公司的催化活性和激活没有(Buechler et al ., 1991)。这个观察是一致的假设两个亚基在协会中发挥作用的血红素酶。定义的角色绑定中的每个单元的血红素,少守恒的n端序列的缺失的α1(删除131残留物)或β1(删除64残留物)进行。删除β-subunit导致损失的敏感性不,确认这个亚基的重要性没有激活的酶(福斯特把et al ., 1996)。删除α1-subunit没有改变无响应的国网公司(福斯特把et al ., 1996)。然而,血红素结合国网公司要求两个完整长度的子单元的存在暗示一个角色在协调α-subunit血红素(石头和Marletta, 1994年)。
半胱氨酸的存在78年和半胱氨酸124年β-subunit很重要的协调国网公司的血红素组(福斯特把et al ., 1996)。这是在协议与观察到的半胱氨酸残基的重要性在其他heme-containing蛋白质。在细胞色素C,血红素共价结合到蛋白质通过硫醚债券两个半胱氨酸残基,符合国网公司的建议血红素也可以通过绑定β-subunit半胱氨酸残基。
在国网公司,每个异质二聚体包含大约一个亲和力不高的血红素(Gerzer et al ., 1981 a)。这与血红素在其他蛋白质,如血红蛋白和肌红蛋白,它具有高度的亲和力在有氧环境中氧和氧结合优先形成ferrous-oxy物种,而不是ferrous-nitrosyl物种。即使在有氧环境,国网公司更喜欢没有绑定而不是氧。如果国网公司结合氧,它形成一个ferrous-oxy物种必须交换氧气,没有形成一个亚铁亚硝酰复杂(Gerzer et al ., 1981 a,b,c)。氧化亚铁血红素组铁状态会导致酶活性的丧失,通常一个完整的损失的血红素蛋白质。铁氰化物氧化国网公司铁状态,这是对任何刺激。因此,减少代理商如硫醇、抗坏血酸盐,或二硫苏糖醇提高酶的激活,可能通过维持铁金属卟啉的亚铁状态是不敏感的。
3所示。调节可溶性Guanylyl环化酶配体。
一氧化氮。
没有是一个自由基,激活国网公司直接绑定到血红素形成ferrous-nitrosyl-heme复杂(图。3)。的半衰期ferrous-nitrosyl血红素4分钟和3 h之间20°C (赴et al ., 1979;沙玛和兰尼,1978)。没有绑定到第六血红素环的位置,打破债券之间的轴向组氨酸和铁,并形成与铁(无花果。3)。这导致5-coordinated戒指,没有现在的排名靠前,位于第五。去除血红素导致的损失没有响应。一氧化碳(CO),国网公司的另一个催化剂,还可以结合国网公司的血红素组,产生一个6-coordinated复杂(图。3)。但是,没有比公司是国网公司的更强有力的催化剂,以及纯化酶被激活100 - 200倍,不,但只有约4倍公司(石头和Marletta, 1994年)。
没有绑定到国网公司似乎适合模型基于两个血红素的数量。小种群,其中包含28%的血红素,最初形成6-coordinate复杂没有然后迅速转换为5-coordinate亚硝酰复杂。没有这个群体分离∼20多岁−1。第二个人口,其中包含72%的血红素,也形成了一个6-coordinate亚硝酰复杂,但转换5-coordinate复杂慢得多。不分离这人口0.1到1.0秒的速度−1太慢了,停用酶。这缓慢的离解的亚铁血红素在第二人口将重新组合的组氨酸在血红素铁。国网公司的二聚状态不受约束的影响没有血红素。5-coordinate含铁酶和5-coordinate亚硝酰酶表现出相同的分子质量的形式∼200 kDa (石头和Marletta, 1996年)。
电子顺磁共振光谱学的国网公司提出一个模型的形成5-coordinated nitrosyl-heme复杂结果打破债券之间的轴向咪唑配体和血红素铁。这将创建一个构象改变蛋白质的结构,激活国网公司。铁是流离失所的从飞机上由于位阻,创建一个类似于原卟啉IX的结构(PPIX)以开放的核心(无花果。3)。开放的中央核心的存在原卟啉环的结构是激活的关键酶(石头et al ., 1995)。
原卟啉IX。
PPIX,血红素的前体,是一种天然的化合物合成的甘氨酸,激活heme-deficient heme-free, heme-containing形式的国网公司以血红素,没有独立的方式。PPIX结合国网公司在低浓度(Kd= 1.4海里),形成复杂的不稳定在凝胶过滤分离或透析(Ignarro et al ., 1982 b;Wolin et al ., 1982)。卟啉环的有两个部分,对绑定的血红素国网公司很重要。两个乙烯基组在位置2和4是必不可少的创建和卟啉环之间的疏水作用的酶。如果这些组织取代乙烯疏水或极性基团,国网公司降低了酶活性由于卟啉环之间的弱相互作用和酶。此外,带负电荷的羧基组位置附近的丙酸残留的6和7形式静电债券基本的残留物,如精氨酸、国网公司的脱辅基蛋白(Ignarro et al ., 1984)。
国网公司的PPIX对动力学参数的影响是类似的。在毫克2 +三磷酸鸟苷,PPIX增加的具体活动heme-containing牛肺国网公司从0.1 - -0.2到2 - 8μmol cGMP /分钟/毫克的蛋白质和降低了K米从100年到56μM。在锰的存在2 +三磷酸鸟苷,PPIX增加特定活动略(从0.3 - -0.6到1 - 1.4μmol cGMP /分钟/毫克蛋白),但是K米保持不变(Ignarro et al ., 1982 b;Wolin et al ., 1982)。PPIX改变酶催化,在某种程度上,通过增加酶的亲和力为毫克2 +三磷酸鸟苷或免费毫克2 +,因为二价阳离子与PPIX容易形成复合物。这PPIX和二价阳离子之间的直接交互激活国网公司的卟啉的关键。有关的酶PPIX结合毫克2 +三磷酸鸟苷或非复杂毫克2 +,但不只是三磷酸鸟苷(Ignarro et al ., 1984)。二价阳离子(毫克2 +、锰2 +),与三磷酸鸟苷是绝对需要复杂衬底代数余子式支持guanylyl环化酶催化活性。
一个铁原子可以合并后,原卟啉合成蛋白质的组成血红素(ferroprotoporphyrin铁2 +)或血色素(ferriprotoporphyrin铁3 +)。尽管血红素所需辅基调解没有国网公司的激活,ferro-PPIX PPIX是竞争性抑制剂(K我= 350海里)(Ignarro 1994)。血色素非竞争性地抑制基底活动在Mg2 +三磷酸鸟苷(K我= 2.8μM)或锰2 +三磷酸鸟苷(K我= 8.3μM)。血色素浓度低于1.5μM竞争性抑制PPIX不改变guanylyl环化酶活动(K我= 0.35μM)。这表明ferroprotoporphyrin和ferriprotoporphyrin争夺相同的结合位点在国网公司(Wolin et al ., 1982)。然而,血卟啉第九,ferriprotoporphyrin乙烯基组与羟乙基两组而不是原卟啉,激活国网公司。血卟啉第九不自然发生,不如PPIX强有力的催化剂。改变PPIX产生类似物的结构,如尿卟啉,粪卟啉,我和二甲基酯PPIX,国网公司活动(没有影响Ignarro et al ., 1982 b)。
催化机理。
催化领域中糖基的α-和C和β-subunits份额显著的同源性1和C2腺苷酸环化酶催化域和催化热解色谱领域。正如上面所讨论的,coexpressionα-和β-subunits形成异质二聚体拥有两个催化领域,需要表达的酶活性。虽然两个催化域存在,每个特定的残基有助于一个衬底绑定和催化网站(刘et al ., 1997 b)(图。1)。二聚作用是由一个特定的地区,包括同源二聚作用域和位于近端催化域的α-和β-subunits(无花果。1)。的确,coexpressionα1和β1拥有c端域的二聚作用和催化区域为基底cGMP足够生产(威德尔et al ., 1995)。核磁共振光谱学的国网公司纯化大鼠肝脏证明cGMP和焦磷酸催化三磷酸鸟苷(唯一的产品蔡et al ., 1980)。分析热解色谱纯化从海胆精子α-phosphoanhydride债券建立网站的乳沟在催化(Walseth et al ., 1981)(图。2)。催化反应的立体化学的课程的考试国网公司纯化使用[α-从牛肺18O]三磷酸鸟苷,cGMP的形成是一个直接置换反应(发送et al ., 1983)(图。2)。
d。二价阳离子。
热解色谱和国网公司都需要二价阳离子作为衬底代数余子式和变构调节剂表达最大催化活性(综述Waldman和Murad, 1987)。他们所有核苷酸环化酶要求嘌呤核苷酸基板与二价阳离子结合形成螯合物的催化域和接受酶环化。锰2 +和毫克2 +最优二价阳离子衬底代数余子式(审查Waldman和Murad, 1987)。使用毫克2 +随着基质阳离子代数余子式,国网公司和基底催化热解色谱展览活动完全由ATP敏感的激活和配体。事实上,毫克2 +可能是生理阳离子支持guanylyl环化酶在体内的活动。相比之下,使用锰2 +随着基质阳离子代数余子式,guanylyl环化酶表现出最大催化活性是由核苷酸对进一步激活和配体(Hardman萨瑟兰,1969;木村et al ., 1976;蔡et al ., 1978;加伯,1979;莱文et al ., 1979;Gerzer et al ., 1981 c;Zwiller et al ., 1981)。这是一个核苷酸环化酶的一般特性。腺苷酸环化酶使用Mn ligand-independent的方式也被激活2 +基质阳离子代数余子式。Mn的确切机制2 +激活guanylyl环化酶ligand-independent方式仍不清楚。然而,它是一种蛋白质,因为Mn的内在特征2 +激活GC-A纯化ligand-independent方式同质性(黄et al ., 1995)。除了作为衬底代数余子式,二价阳离子也激活guanylyl环化酶的变构方式(加伯和灰色,1974年;Waldman和Murad, 1987)。事实上,二价阳离子浓度超过核苷酸衬底需要最大的催化活性。二价阳离子的精确分子机制变构调节guanylyl环化酶仍不明朗。
钙还支持guanylyl环化酶催化活性衬底代数余子式,但似乎是国网公司的负变构调制器(莱文et al ., 1979)。事实上,Ca2 +在一些生理系统和cGMP敌对的功能。在血管平滑肌收缩是由(Ca的增加2 +]我,而松弛的发生是由于增加(cGMP)我。光明在视网膜感光细胞,减少(cGMP)我和减少(Ca2 +]我,而增加(cGMP)我和[Ca2 +]我与黑暗光照后恢复。然而,它已经很难检查监管guanylyl酸环化酶的Ca2 +因为calcium-dependent监管机制存在的外在guanylyl环化酶在细胞。例如,Ca2 +与GCAPs调节视网膜guanylyl环化酶,Ca2 +是不需要代数余子式合酶(NOS),其产品是国网公司的强有力的催化剂(Yarfitz赫尔利,1994年;Yu et al ., 1999)。最近,Ca2 +国网公司的监管,研究了在不同的系统中其他的Ca2 +端依赖监管机制缺席(帕金森et al ., 1999)。人类胚胎肾细胞(HEK 293),不表达内源性NOS和表达量较低的国网公司活动,与α1 cotransfected和β1鼠国网公司的子单元。Ca2 +抑制基底和NO-stimulated原油和immunopurified国网公司准备从这些细胞。三磷酸鸟苷的生理范围,Ca2 +抑制浓度,和鸟嘌呤nucleotide-dependentK我= 2.6μM。抑制国网公司是由细胞外钙2 +和释放(Ca2 +]我池通过受体介导机制。Ca2 +减少两个V马克斯和K米国网公司,符合一个竞争力Ca的抑制机理2 +相互作用与衬底(毫克2 +三磷酸鸟苷)或一个产品(cGMP或焦磷酸(PPi)]催化部位。这种机制是类似于p区抑制腺苷酸环化酶,其中嘌呤核苷酸结合抑制酶活性的催化装置的产品毫克2 +ppi (德绍,1997)。这项研究表明国网公司可能作为一个特定的传感器(Ca2 +]我产生相互协调的监管(Ca2 +]我和(cGMP)我。
三世。环磷鸟苷和细胞信号
答:介绍
内源性和外源性的化合物,包括内分泌物、激素、神经递质,通过cGMP和毒素,产生细胞反应。这些反应包括合成的生化机制(guanylyl酸环化酶,见上图),针对(繁多,见下文),和退化(cGMP的pd,见下文)。cGMP的特异性的细胞反应是由cGMP-binding图案在目标蛋白质。两个进化截然不同的变构网站绑定cGMP存在于真核细胞。一个发生显著的序列同源性在包裹和cAMP-dependent蛋白激酶(PKA)和循环nucleotide-gated (CNG)阳离子通道,而另一个发生在cGMP-regulated pde。此外,增加的结果(cGMP)我是由目标蛋白和基质的类型和组合,cGMP-metabolizing酶表达的细胞,其细胞内colocalization和组织为选择性隔间和细胞器。例如,受磷和IP3肌浆网中受体基质包裹和PKA,但colocalization激酶及其基质的模式根据细胞类型不同。与这两种基质包裹colocalizes smc,而PKA colocalizes心肌细胞动作电位与他们(林肯et al ., 1995)。因此,蛋白的磷酸化受和IP3受体在smc有助于放松反应,cGMP-dependent。相比之下,发生心肌细胞动作电位的磷酸化底物通过营地/ PKA通路,促进Ca2 +封存和缩短心脏收缩。最后,cGMP生理和病理生理条件下可能有不同的影响。例如,在激活中性粒细胞,cGMP / PKG磷酸化酶的中间丝波形蛋白在瞬态colocalization灯丝。在静止的中性粒细胞,cGMP类似物不产生波形蛋白的磷酸化(怀亚特et al ., 1991)。
以下部分提供了一个简短的角度如何guanylyl环化酶和cGMP融入跨膜信号级联,一个广泛的概述事件下游调节guanylyl环化酶和(cGMP)我胎盘哺乳动物系统中积累。出于这个原因,cGMP的生理作用在特定的细胞系统中,如人类血液细胞,在原始的单细胞生物。感兴趣的读者,杰出的评论是可用的,特别关注PKA和包裹(弗朗西斯和卡宾,1999年),CNG阳离子通道(考普1995;齐默尔曼,1995;比尔et al ., 1999 b),cGMP-regulated pd (Beavo 1995;Juilfs et al ., 1999)。此外,一个完整的纲要cGMP的功能是可用的(1994年的Murad)。
b蛋白激酶
1。循环GMP-Dependent蛋白激酶。
包裹代表的主要细胞内中介cGMP的信号。Ligand-induced海拔(cGMP)我诱发binding-dependent激活的包裹导致催化ATP的γ-phosphate转移到目标蛋白质的丝氨酸或苏氨酸残基。这种磷酸化蛋白介导细胞外刺激的翻译成一个特定的生物功能。
两种不同的包裹已确定的基因在哺乳动物。一个基因位于人类10号染色体编码Iα和Iβ亚型的包裹我,来自可变剪接的氨基端区域(田村et al ., 1996 a)。另一个是位于人类染色体4和编码PKG二世(Orstavik et al ., 1996)。包裹我是胞质76 kda为哺乳动物组织中广泛表达,特别是在小脑,血小板和平滑肌(罗曼et al ., 1997)。不同的n端结构域之间的两个包裹亚型赋予不同的绑定cGMP的亲和力。PKG Iα高和低亲和力结合位点显示积极的合作行为。PKG Iβ有两个cGMP结合位点的特征是低亲和力和协调(Pfeifer et al ., 1999)。此外,尽管这两种亚型的表达已经检测到同样的人体组织,包裹Iα检测主要在血管系统、肾、肾上腺、而只有包裹Iβ检测子宫(田村et al ., 1996 a)。
包裹二世是一个86 kda膜结合为。在心血管系统,丰富的大脑和肠道,也表现在肺、肾、骨(Uhler 1993;Jarchau et al ., 1994;罗曼et al ., 1997)。包裹二世的氨基酸序列不同于包裹我主要在N末端的序列;该地区独特的网站直接酶的胞内定位。包裹2包含了一个myristoylated膜协会需要的网站,而包裹我包含一个乙酰化网站(罗曼et al ., 1997)。主要区别cGMP结合位点的两个包裹,包裹二世有最小的亲和力和协调。另一个区别是很大的分歧他们的底物选择性,体内更明显(Pfeifer et al ., 1999)。此外,两种形式的包裹在不同的细胞表达,除了在胫骨生长板软骨细胞的新生鼠(Pfeifer et al ., 1996)。
所有已知的包裹是由氨基端、监管和催化领域。n端结构域包含五个监管网站:(1)亚基二聚网站组成的一个α-helix守恒的亮氨酸、异亮氨酸七个重复;(2)autoinhibitory网站,参与抑制催化域的cGMP的缺失;(3)自身磷酸化网站,cGMP的存在会增加基底催化活性和pkg为阵营的亲和力;(4)规范的网站亲和力和cGMP结合位点的合作行为;(5)细胞内定位的网站,确定酶的相互作用与特定亚细胞结构。监管域包含两个环核苷酸结合位点(通常称为“A”和“B”)后,允许充分激活酶特定的绑定cGMP的两个分子。最后,催化领域,位于pkg的糖基,包含Mg的结合位点2 +atp和目标蛋白(林肯et al ., 1995;罗曼et al ., 1997;Pfeifer et al ., 1999)。
广泛的蛋白质磷酸化通过体外包裹,但只有少数的磷酸化已经演示了体内。这种明显的差异可能用这一事实来解释,除了衬底的识别序列,在生理条件下包裹的底物特异性是由细胞内的空间定位。这导致了一个独特的高分子聚合enzyme-target蛋白质组成的复杂。
当前工作假说表明,包裹我作为可溶性胞内的调制器(Ca2 +]我第二,而包裹调节流体在细胞膜内稳态。生物基质包裹我在概念上可以细分为三类,“古典”,“新”和“假设”的目标。“古典”的目标显然是公认为基质,在体外或体内,被包裹我,磷酸化和有良好功能。这一组包括:(1)的IP3受体和受主要涉及SMC放松(Raeymaekers et al ., 1990;Komalavilas和林肯,1996年);(2)vasodilator-stimulated磷酸化蛋白和波形蛋白,分别参与血小板和中性粒细胞激活(Pryzwansky et al ., 1995;Aszodi et al ., 1999);(3)G衬底,强烈表达小脑浦肯野细胞,它作为一种磷酸酶抑制剂(Endo et al ., 1999);(4)凝血恶烷2受体的激活被发现PKG-mediated磷酸化的抑制血小板(王et al ., 1998)。“新”目标蛋白质基质包裹我最近被描述或相互矛盾的证据对其磷酸化包裹。这组包括:(1)l型钙2 +通道和Ca2 +激活K+通道,经磷酸化,导致血管平滑肌张力的规定和心脏收缩性(扬et al ., 1988;深尾三硕et al ., 1999);(2)Ca2 +端依赖胞质磷脂酶一2,与肠道平滑肌松弛(没吃,Makhlouf 1998);(3)酪氨酸羟化酶的活性在完整的观察牛嗜铬细胞增加PKG I-mediated磷酸化(Rodrı́guez-Pascual et al ., 1999);和(4)肌球蛋白轻链磷酸酶的myosin-binding亚基,介导SMC放松和血管舒张(Surks et al ., 1999)。大多数,如果不是全部,这些底物磷酸化的亚型Iα包裹,已经提出“假设”的目标蛋白质,但不证明,被包裹Iβ磷酸化。这组包含假定的基质的基础上预测实验演示cGMP / PKG我介导的过程。例如,细胞骨架和收缩蛋白(即。,米yosin light chain, calponin, desmin, connexins) are thought to be PKG Iβ target molecules in the regulation of vascular remodeling and neoangiogenesis (林肯et al ., 1998;Eigenthaler et al ., 1999)。同样,突触囊泡蛋白(即。,rabphilin-3A) may be phosphorylated by PKG I and mediate synaptic plasticity and neurotransmission (钱et al ., 1996;灰色et al ., 1999;Yawo 1999)。cGMP / PKG我途径涉及基因表达的启动子的控制响应元素(即。、血清反应元素,AP-1结合位点,营地响应元素)(Gudi et al ., 1996,1997年)。
与包裹我,唯一公认的“古典”磷酸化的底物包裹II是囊性纤维化跨膜电导调节(雌性生殖道)在肠粘膜细胞(Vaandrager et al ., 1997)。本地化的PKG II的肠上皮细胞顶膜的小肠CFTR允许它使磷酸化检测响应GC-C-induced cGMP的形成。CFTR的磷酸化检测线圈产生电致氯电流和随后的水在小肠分泌。Colocalization coregulation PKG II的表达和氯通道在大鼠肾内髓质建议类似的机制可能调节肾脏功能(Gambaryan et al ., 1996)。包裹二世也可能控制肾素系统和骨软骨内骨化和增长(Pfeifer et al ., 1996;瓦格纳et al ., 1998)。然而,目标分子在这些后者过程是未知的。
2。循环AMP-Dependent蛋白激酶和环磷鸟苷信号。
因为pka包含特定的环核苷酸结合域与显著的同源性包裹,他们可能会激活cGMP,尽管50倍选择性低于阵营。尽管pka的核苷酸结合位点和包裹是同源(Pfeifer et al ., 1999),这些网站存在差异,特别是在关键氨基酸残基的取代(罗曼et al ., 1997)。
有一些争论关于交错活化蛋白激酶的环核苷酸。有趣的是,许多认识生理基质包裹也为pka基质。此外,cGMP似乎一致行动,在多种细胞过程。因此,包裹和PKA抑制IP3端依赖释放Ca2 +在分散和诱导放松兔和豚鼠胃肌肉细胞(没吃,Makhlouf 1995)。类似地,异丙肾上腺素或SNP诱导胞质Ca的磷酸化2 +端依赖磷脂酶一2分别通过激活PKA或包裹在smc兔小肠纵行肌层(没吃,Makhlouf 1998)。同样的效果是通过同时刺激的激酶通过贵宾和异丙肾上腺素,导致这些细胞的放松反应。
在脊椎动物,cGMP和营地放松血管平滑肌,抑制血小板激活和调节氯和水在小肠分泌。环腺苷酸cross-activates包裹在不同血管组织,包括大鼠主动脉和猪冠状动脉(江et al ., 1992;Eckly-Michel et al ., 1997)。体外,cGMP cross-activated PKA,调节分泌的液体由圣在人类肠道细胞(福特et al ., 1992;曹国伟et al ., 1994)。功能融合cGMP和营地也可能发生在下游水平,比如在目标蛋白激酶的水平。基于底物磷酸化PKA和包裹的身份,有大量重叠的两个环核苷酸的能力管理有限公司2全身cerebrovasodilatation的成年大鼠软膜的动脉(王et al ., 1999)。这些激酶可能采取合作态度和磷酸化的活动需要一个全面的影响。号的过度表达,导致没有水平的提高,导致交错活化PKA的大量增加cGMP (林肯et al ., 1995)。这种机制介导的抑制SMC增殖和PKG Iα表达在大鼠主动脉和基础nitrovasodilator宽容的发展牛,鼠主动脉SMC不断暴露在这些代理(康威尔et al ., 1994;索夫et al ., 1997;林肯et al ., 1998)。
尽管上述体外观察表明cGMP可以通过PKA体内信号,其他的考虑不支持这一假说。完整的细胞表现出广泛的亚细胞划分范围第二信使,酶及其底物有限的“工作单位”和代表交错活化的一个障碍。此外,PKA和包裹必须锚到特定的细胞内蛋白质来执行他们的生理功能。因此,只有PKA Iα,但不是PKA IIα,重新分配与t细胞受体与复杂在anti-CD3诱导早期激活和限制。PKA Iα从而介导的抑制t细胞的增殖(Skalhegg et al ., 1994)。同样,包裹瞬变与磷酸化,波形蛋白在人类中性粒细胞激活A23187,但不是在静止细胞(Pryzwansky et al ., 1995)。Colocalization激酶和基质,由目标蛋白质,现在被认为是特异性的主要决定因素的影响(Pfeifer et al ., 1999)。
生理相关蛋白激酶的cross-regulation环核苷酸被研究的挑战与纯合子小鼠包裹我——或者包裹II-null突变。循环GMP-induced放松在主动脉环或胃底肌肉条准备包裹I-deficient小鼠受损,而cAMP-induced放松不是(Pfeifer et al ., 1998)。这些突变小鼠是高血压和缺乏常规的肠道蠕动,说明包裹我是cGMP的特定中介效应在平滑肌,体内。此外,有一个有缺陷的cGMP-mediated抑制血小板的活化反应突变小鼠,而cAMP-mediated抑制不受损(Massberg et al ., 1999)。的确,没有交错活化的血小板PKA cGMP观察。最后,与纯合子小鼠PKG II零变异显示选择性降低ST -和8-bromo-cGMP-induced肠道分泌(Pfeifer et al ., 1996)。相比之下,从小肠引起电致离子分泌阵营类似物并不是突变的影响。
这些观察表明,体内,cGMP和营地信号级联引起各种重要生理效应很大程度上独立。不管是cGMP激活PKA还是营激活PKG可以完全弥补其他的核苷酸。虽然直接cross-regulation蛋白激酶的环核苷酸可能不似乎是一个占主导地位的过程调节信号,有证据表明,cGMP调节电致氯化物分泌小和大肠通过抑制PDE的3型异构体,增加细胞内营,激活PKA (Vaandrager et al ., 2000)。这些数据将更详细地讨论在本文后面的部分。
c循环Nucleotide-Gated渠道
CNG通道是一个家庭的各种细胞电压门控阳离子通道。天然气管道的特征包括:(1)六个跨膜段的存在(S1-S6),(2)之间的ion-conducting孔隙S5和S6地区,和(3)的两个互动细胞质膜的监管领域,由N和C末端的通道。此外,CNG通道响应特定的环核苷酸控制循环nucleotide-binding域的C末端循环nucleotide-dependent同源的蛋白激酶(比尔et al ., 1999 c)。他们选择性敏感环核苷酸的八个β-rolls和三个α-helices驻留在C末端,pka和包裹。因此,尽管所有天然气通道被激活cGMP和营地,某些同形像更敏感比营地cGMP,反之亦然。
Hyperpolarization-activated循环nucleotide-gated (HCN)通道特定亚科的CNG频道。这些渠道表达是通过控制调节大脑和心脏和环核苷酸的直接绑定和质膜超极化(路德维希et al ., 1998,1999年)。三个不同的HCN通道(HCN 1 - 3)已经从老鼠的大脑,克隆和公认的第四个成员也被报道(McCoy et al ., 1995)。这些通道具有独特的结构特点在S4孔隙地区和跨膜段赋予选择性离子通透性和独特的电压灵敏度,分别。环磷鸟苷增强当前的振幅和通道的激活率从HEK 293年膜细胞表达HCN2瞬变,和营地有一个更大的效果(路德维希et al ., 1998)。HCN2透水Na+和K+和演示功能性质相似的离子电流的起搏细胞在中枢神经系统和心脏。此外,两个截然不同的人类基因编码表达的克隆人类心脏和HCN通道HEK 293个细胞电生理特性兼容一个假定的函数作为效应器心脏起搏的体内(路德维希et al ., 1999)。
天然气的主要家庭频道调节流入Na+和Ca2 +进入细胞,是由四聚物的蛋白质,由环核苷酸直接“打开”。相比HCN, CNG蛋白质显示非常微弱的电压灵敏度和由α-β-subunits heterotetrameric组合。只有α-subunits形式功能单体的渠道表达在不同的系统(比尔et al ., 1999 c)。三个高度同源α-subunits (CNG 1 - 3),最初从棒克隆,锥,嗅觉神经元,被发现在胰腺,脾脏、睾丸、卵巢、肾、肺、脑、心脏、肾上腺和肠(McCoy et al ., 1995)。CNG存在通道被发现在老鼠的舌头的味蕾感觉器官和可能是至关重要的分子(Misaka et al ., 1997)。只有两个β-subunits (CNG4和CNG5)已确定(比尔et al ., 1999 c)。CNG 4基因拼接不同,导致一些特异性亚型(比尔et al ., 1999 c)。本机CNG渠道被认为是由α-和β-subunits虽然homotetramersα4和β4可能存在。特定的结构特点具有离子选择性渗透孔隙的CNG地区渠道。所有通道进行单价阳离子但更渗透Ca2 +比娜+在生理条件下(细胞外钙2 +>其他单价阳离子)。此外,钙2 +调节通道从额外的或细胞内舱压敏电阻器的方式(比尔et al ., 1999 c)。Ca2 +块频道活动直接与高亲和力结合位点和间接交互通过激活其他蛋白质,如钙调蛋白。后者效果嗅觉通道活动的一个重要组成部分。
有人建议,β-subunits充当CNG渠道活动的内部监管机构,赋予特定的属性,如单通道“闪烁”和亲和力营(比尔et al ., 1999 c)。天然气管道的其他调节地区包括氨基之间的链接器肽段和S6跨膜域和胞质循环nucleotide-binding域。这些确定受体激动剂的亲和力和通道控制和基础cGMP法规在视杆细胞和视锥细胞的视觉传导嗅觉和营地的监管(比尔et al ., 1999 c)。
环磷鸟苷介导phototransduction的视杆细胞和视锥细胞和视网膜内调节神经传递。这些过程取决于天然气通道和高(cGMP)我穆勒在神经节、双极和胶质细胞。事实上,光感受器代表的主要细胞类型展示cGMP / CNG效果。删除CNG 3在老鼠身上导致选择性破坏cone-mediated光敏反应,特点是累进的锥体光感受器变性,但不是棒或任何其他视网膜细胞类型(比尔et al ., 1999))。这些老鼠是肥沃,正常发育。这些数据支持了假设选择性突变在人类CNG 3基因构成全色盲,一种人类疾病,其特征是全色盲(比尔et al ., 1999))。
d .循环GMP-Regulated磷酸二酯酶
描述的生化、药理和结构概要文件已经确定至少10个不同基因家族pde的哺乳动物。每个成员都包含一个守恒的催化域∼270个氨基酸的糖。这个域劈开磷酸二酯键,水解3′,5′环核苷酸对其相应的核苷酸,5′一磷酸(McAllister-Lucas et al ., 1995)。所有pde包含异构监管领域和功能为二聚体,尽管后者的功能意义功能仍是未定义的。
PDE家庭1、2、3和10水解cGMP和营地;PDE家庭4、7和8优先裂开营地;和PDE家庭5、6和9水解cGMP。pde活性的细胞信号是至关重要的,因为环核苷酸代谢调节细胞内的浓度,影响后续的细胞和行为反应。pd调节心脏功能、肾上腺类固醇生成,男性勃起反应,phototransduction (Juilfs et al ., 1999)。具体PDE目标站点可以本地化酶在靠近选择蛋白质,从而调节环核苷酸水平在特定的隔间。
环磷鸟苷调节pde通过三种不同机制:(1)增加活动通过群众运动(PDE5、6和9),(2)改变阵营的水解率通过竞争在催化部位(PDE1、2和3),和(3)通过直接绑定到特定的变构调节酶活性的网站(PDE2 5、6和10)(卡宾和弗朗西斯,1999)。
非催化的变构网站允许cGMP调节pde的活动在几个方面。PDE2 cGMP绑定域名,5、6、10包含两个同时同源网站约110位于N末端氨基酸。它们含有一种氨基酸序列不同于循环nucleotide-dependent激酶和CNG频道,代表一个单独的类cGMP-regulated蛋白(卡宾和弗朗西斯,1999)。PDE10催化环核苷酸的水解,但其生理功能尚不清楚。PDE2广泛分布,是为100 - 150 kda的子单元,和催化的水解cGMP阵营。环磷鸟苷结合变构网站,刺激PDE2活动,增加环鸟苷酸水解,形成负反馈调节机制(cGMP)我。同样,cGMP提高PDE2-mediated退化的阵营,因此cross-regulating其细胞内浓度(McAllister-Lucas et al ., 1993)。
93 - kda PDE5,为子单元,特别是降低环鸟苷酸。直接绑定cGMP变构网站促进磷酸化PDE5的包裹或PKA,从而间接地刺激酶活性。有人建议,绑定cGMP的变构直接激活PDE5网站,但这种效应还没有被证明(卡宾和弗朗西斯,1999)。最后,PDE6杆(PDE6-A和- b)和锥(PDE6-C)感光细胞是由两家大型催化地活动单元(α,β棒;α′2在锥)与各种小抑制γ-subunits和监管δ-subunit。相信cGMP绑定的变构网站PDE6调节催化亚基之间的相互作用,抑制子单元,和transducin phototransduction(迈出的重要一步McAllister-Lucas et al ., 1993,1995年)。
e环磷鸟苷和细胞生理学
1。血管平滑肌的蠕动。
血管平滑肌的收缩机制是基于协同和敌对的力量调节(Ca2 +]我。收缩力量提升(Ca2 +]我和/或使细胞内环境2 +,而扩张力量减少(Ca2 +]我和/或降低Ca的细胞内环境2 +。这些对立的过程的影响是磷酸化20-kDa的肌球蛋白轻链(MLC20)在丝氨酸19日由肌球蛋白轻链激酶产生血管收缩,由肌球蛋白轻链和脱磷酸作用MLC20磷酸酶产生血管舒张,分别为(班纳特和沃尔德曼,1995)。
外源性和内源性化合物(如nitrovasodilators endothelium-derived放松的因素,和利钠肽)产生通过增加血管舒张(cGMP)我。环磷鸟苷放松血管smc的减感作用的Ca的伸缩装置2 +和降低(Ca2 +]我(无花果。4)。三硝酸甘油酯或SNP-mediated血管放松没有显著减少(Ca2 +]我据报道在猪颈动脉和冠状动脉(安倍et al ., 1990;麦克丹尼尔et al ., 1992)。环磷鸟苷诱导转向右边的主成分分析2 +张力曲线在大鼠肠系膜动脉permeabilizedα-toxin和耗尽(Ca的商店2 +]我(Nishimura和van Breemen, 1989年)。在类似的实验中与de-endothelized从兔股动脉平滑肌条,8-bromo-cGMP高架ED50Ca的2 +收缩力和MLC20通过间接激活肌球蛋白轻链磷酸化磷酸酶(李et al ., 1997)。

分子机制血管平滑肌放松由环磷鸟苷。环磷鸟苷诱导平滑肌松弛减少(Ca2 +]我并使不敏感的伸缩装置2 +。环磷鸟苷降低(Ca2 +]我(1)抑制Ca2 +通过l型钙流入2 +渠道;(2)增加Ca2 +射流通过激活(2 d) Ca2 +天然气atp酶和(2 b) Na+/ Ca2 +换热器;此外,cGMP可能产生膜超极化激活的(2 c) Na+/ K+atp酶和(2)K+渠道,从而增加Ca2 +Na挤压的+/ Ca2 +换热器;(3)增加的Ca2 +通过激活肌浆网钙封存2 +天然气腺苷三磷酸酶(Ph值,受);(4)减少的Ca2 +通过抑制动员agonist-induced IP3形成或抑制肌浆网IP3受体。R受体;G, G蛋白;PLC,磷脂酶C;IP3R IP3受体。环磷鸟苷逐渐脱敏Ca的伸缩装置2 +(5)可能通过激活肌球蛋白轻链磷酸酶,导致脱磷酸化的20个kDa肌球蛋白轻链。
虽然包裹我已被建议作为校长候选人调解这些cGMP效应,其他分子目标不能被排除在考虑之外。在这其中,PKA是一个可能的候选人,根据生化和功能融合机制cGMP和cAMP-induced血管松弛。然而,如上所述,这个假设的环核苷酸相声似乎并不发生在心血管系统,通过cAMP-independent体内,cGMP信号通路。
环磷鸟苷介导血管平滑肌放松主要是通过降低(Ca2 +]我(无花果。4)。根据组织、物种和细胞基因型和表现型,cGMP可能影响(Ca2 +]我在四个不同的方面:(1)通过减少Ca2 +通过增加Ca涌入,(2)2 +流出,(3)通过促进Ca2 +封存的肌浆网,(4)通过减少Ca2 +动员。因此,cGMP / PKG-dependent机制抑制l型压敏电阻器激活Ca2 +渠道渠道活动的直接损伤和间接超极化的SMC表面通过开放的概率增加KCa通道(阿切尔et al ., 1994;载体et al ., 1997;刘et al ., 1997 a;Ruiz-Velasco et al ., 1998)。包裹的磷酸化假定的receptor-operated Ca2 +α-subunit的渠道和大型电导KCa渠道已经证明,强烈支持减少cGMP-mediated Ca的生理作用2 +涌入(布雷勒et al ., 1991;深尾三硕et al ., 1999)。
两种不同的离子通道的激活,质膜2 +天然气atp酶和Na+/ Ca2 +换热器,可以调解的流出增加Ca2 +从血管smc。挤压的Ca的驱动力2 +从细胞到Na+/ Ca2 +换热器,反过来,可能依赖于其他两个效果由cGMP,细胞内钠的损耗+通过激活钠+- k+通过激活腺苷三磷酸酶和细胞膜超极化K+频道。Ca2 +天然气atp酶和Na+- k+在质膜atp酶活性通过cGMP包裹(吉田et al ., 1992;Tamaoki et al ., 1997)。如上所述,cGMP也可以间接地促进Ca2 +运输通过激活细胞的K+渠道和由此产生的超极化。此外,8-bromo cGMP可能刺激Na+/ Ca2 +换热器在膜电位(独立于任何修改古河道et al ., 1991)。
环磷鸟苷诱导钙的吸收2 +进入细胞内的商店通过激活肌浆网的钙2 +天然气atp酶(Andriantsitohaina et al ., 1995)。背后的分子机制这种效应似乎包裹蛋白的磷酸化受在培养大鼠心肌细胞(Sabine et al ., 1995)。最后,似乎cGMP可能抑制的IP3信号转导通路,从而降低(Ca2 +]我。事实上,cGMP块agonist-induced IP3形成和诱发PKG-mediated磷酸化的IP3受体的肌浆网,随后衰减动员的Ca2 +(藤井裕久et al ., 1986;露丝et al ., 1993;Komalavilas和林肯,1994年,1996年)。
总之,有证据表明一个复杂的角色cGMP在血管平滑肌放松施加的控制(Ca2 +]我(无花果。4)。很可能,体内,不同机制实现协同运作的低水平的(Ca2 +]我和诱导血管放松利用cGMP作为第二信使。
2。肠道液体和电解质内稳态。
圣引起分泌性腹泻通过激活guanylyl环化酶和增加(cGMP)我(无花果。5)。细胞病理效应的目标是肠上皮细胞衬,唯一发现guanylyl环化酶异构体是GC-C圣受体(Vaandrager·德容,1994)。破坏老鼠的基因编码GC-C ST-induced腹泻,导致阻力所需证明GC-C绝对ST-induced肠道分泌(曼et al ., 1997;舒尔茨et al ., 1997)。在动物模型中,绑定的圣GC-C刺激肠道分泌通过增加cGMP,和圣对肠道分泌的影响被cell-permeant模仿cGMP的类似物(场et al ., 1978;休斯et al ., 1978;Mezoff et al ., 1992)。高亲和力的受体圣本地化在肠上皮细胞刷状缘膜从十二指肠到直肠,在小肠密度最高(Krause et al ., 1994 b)。

由圣和GC-C调节肠道分泌。细菌,如大肠杆菌,含有质粒编码同源肽家族的成员STs殖民小肠后食用被污染的食物或水。细菌殖民化导致圣在肠道内腔的生产,专门识别和结合的细胞外领域GC-C,表现在肠粘膜细胞的刷状缘膜从十二指肠到直肠。交互GC-C ST和细胞外领域的翻译在质膜进入细胞质催化域的激活导致的生产和积累(cGMP)我。这个环核苷酸结合并激活包裹二世,也在肠道细胞刷状缘膜局部。cGMP可能激活PKA,直接或通过抑制cAMP-specific PDE和诱导营地的积累。与GC-C和包裹的雌性生殖道二世在刷状缘膜蛋白激酶和PKA生长的基质。雌性生殖道是一个氯通道,其磷酸化PKA或包裹导致持续性开放状态,允许氯向下流动的浓度梯度从细胞到细胞外室。其他离子通道和转运蛋白的细胞保持电中性ST-induced氯流出。从基底外侧Vectoral水通量顶端表面是由这些离子电导,导致液体和电解质的积累在肠道内腔和分泌性腹泻。
雌性生殖道是一个关键的组件调停圣的产肠毒素的效果(图5)。CFTR缺乏功能性检测发生在囊性纤维化患者或与零CFTR突变的检测小鼠圣和cGMP类似物未能引起腹泻。此外,肠道、肺、胰腺发展严重异常水和盐的规定内容(昆廷,1990)。雌性生殖道被公认为环核苷酸的主要中介调节液体和电解质运输在多种上皮细胞,进行整个顶端细胞膜氯电流。除了雌性生殖道,最近的证据显示的作用抑制刷状缘膜电中性的钠吸收,可能由Na+/小时+换热器,在机制ST-induced液体和电解质的分泌(Vaandrager et al ., 2000)。
氯化环磷鸟苷激活雌性生殖道和促进流出,这可能导致肠道水运输到腔(无花果。5)。包裹似乎cGMP的主要分子目标的信号序列导致雌性生殖道激活。CFTR PKG Iα和PKG II使磷酸化检测体外,类似的动力学,认为缺乏具体PKG-mediated函数在这个过程中(法国et al ., 1995)。然而,第二包裹,但不是包裹Iα,colocalizes GC-C在刷边界切除肠上皮细胞,激活雌性生殖道的膜各种细胞系转染与雌性生殖道的补丁(罗曼et al ., 1997)。CFTR Cotransfection鼠肠细胞的检测和PKG II导致雌性生殖道的激活,而Cotransfection PKG Iβ不激活通道(Vaandrager et al ., 1998)。突变的包裹二氨基端myristoylation网站减少了本地化的酶膜和雌性生殖道的损害激活。相比之下,可溶性PKG Iβ嵌合构造和membrane-directed包裹二世的n端结构域获得激活雌性生殖道的能力。意义,代理,增加cGMP PKG II-deficient小鼠小肠的抑制分泌或电致氯电流的感应,而cAMP-induced肠道分泌没有影响(Pfeifer et al ., 1996;Vaandrager et al ., 2000)。这些研究表明,包裹二世是一个主要的雌性生殖道的生理中介在小肠和定位激酶激活细胞膜是其功能在完整细胞的主要决定因素。
有趣的是,除了第二包裹,其他机制似乎调解激活cGMP雌性生殖道的小肠和结肠(无花果。5)。环磷鸟苷可以直接激活PKA,刺激CFTR-mediated氯电流在人类结肠细胞不表达PKG二世(福特et al ., 1992;曹国伟et al ., 1994)。同时,圣诱发电致结肠癌和空肠的氯化物分泌PKG II-deficient老鼠(Vaandrager et al ., 2000)。圣的影响在包裹II-deficient小鼠肠道阴离子分泌被添加氨力农强,PDE3抑制剂的同种型催化降解的阵营,但被cGMP (Vaandrager et al ., 2000)。综上所述,这些数据表明,增加(cGMP)我刺激雌性生殖道的圣可能诱发磷酸化和电致氯运输通过激活第二包裹或PKA。后者激活蛋白激酶可能直接由cGMP或当地积累(营)我反映抑制PDE3的增加(cGMP)我(无花果。5)。
一个关键问题有关cGMP-mediated调节肠道液体和电解质交通的内源性配体激活GC-C的身份。存在一个唯一的悖论哺乳动物细菌性肠毒素受体被证明解决提取大鼠的肠道刺激GC-C T84人类肠道细胞,增加环鸟苷酸浓度(Currie et al ., 1992)。guanylin生物活性物质,是一种15-residue肽结构上和功能上同源圣guanylin是由上皮细胞或细胞控制小肠上皮功能、结肠癌、肾上腺、子宫、肾、胰腺、输卵管。在人类肠道细胞,T84 guanylin刺激cGMP合成能力降低了10倍与圣(的强项和库里,1995)。Uroguanylin, guanylin-like肽隔绝负鼠和人类的尿液,刺激对大鼠结肠GC-C,体外,T84细胞,尽管比圣效力较低(哈姆拉et al ., 1993;北城et al ., 1994)。有趣的是,影响T84细胞强烈影响细胞外pH值:在低pH值(即。,5。0–5.5) uroguanylin is 100-fold more potent than guanylin, whereas at a pH of 8.0, guanylin is 4-fold more potent than uroguanylin (哈姆拉et al ., 1997)。后发现的可能性的节段性监管这些内源性肽肠,因为肠道流明的pH值变化明显从胃到直肠。
信号级联由guanylin / uroguanylin肠圣(图和描述是一样的。5)。GC-C绑定后,在肠上皮细胞顶膜,配体刺激内在guanylyl环化酶催化活性,启动一个级联有(1)积累(cGMP)我,(2)刺激的膜相关包裹II和/或PKA,和(3)磷酸化的雌性生殖道(Gudi et al ., 1996)。电致氯电流,反映出雌性生殖道的激活,导致净分泌盐和水进入肠道流明。已经提出,生理浓度的guanylin可以作为肠道液体传感器,防止过度脱水和净化肠道粘膜(罗曼et al ., 1997)。然而,在(即含量异常地高。,在pathological states), guanylin may cause an exaggerated loss of fluid and diarrhea (的强项和库里,1995)。
水和离子运动的模式由guanylin,体内,最近研究在大鼠肠道循环(关闭会飞的et al ., 1997)。Guanylin(2μM)刺激分泌的水,Na+,Cl−在十二指肠、回肠和结肠通过抑制Na+氯的吸收和刺激−流出。这种影响是低于圣各肠段。相比之下,没有检测到电解液通量观测空肠。然而,在随后的实验中麻醉大鼠空肠的封闭循环,老鼠和人类guanylin(1μM)抑制吸收液体和氯化钠(理念et al ., 1999)。在同样的实验中,guanylin uroguanylin,圣诱导等渗液体运动进入空肠的腔,但只有圣和uroguanylin增加了通过刺激碳酸氢盐分泌腔的pH值。
虽然上面的讨论表明,cGMP参与肠道液体和电解质内稳态的规定,这种机制的生理意义尚不清楚。因此,存在GC-C爬行动物和鸟类的肠子表明一个明确的进化保存的同种型guanylyl环化酶(Krause et al ., 1995,1997年)。然而,老鼠GC-C或其下游基因的分子目标,包裹II,中断没有在他们的肠道发育和功能明显异常,除了guanylin / uroguanylin /圣没有反应(舒尔茨et al ., 1997;促进et al ., 1999;Pfeifer et al ., 1999)。
3所示。Phototransduction。
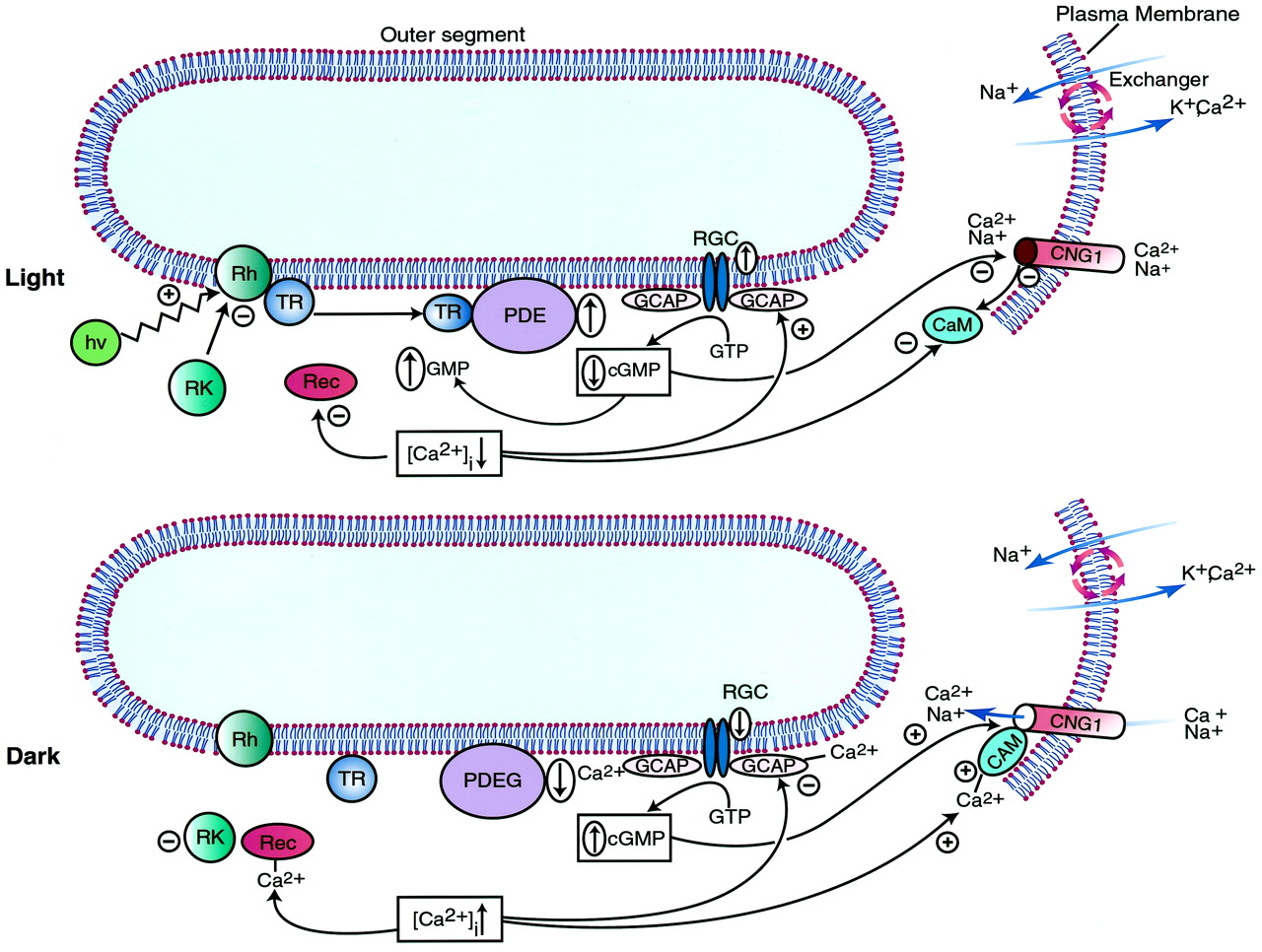
Phototransduction视网膜外节的代表一个生动的例子,cGMP在生理过程的重要性。的确,cGMP调节视觉激发的复苏阶段和适应背景光(艾姆斯et al ., 1999)。视杆细胞和视锥细胞都含有独特的蛋白质合作采取行动控制键第二信使(cGMP)我和[Ca2 +]我。这些,反过来,调节整个机制phototransduction并确定生理反应。
视网膜细胞含有两种亚型的膜结合guanylyl环化酶,GC-E和- f。这些亚型表达只在脊椎动物的感光细胞,激活了它们形成为互动与特定的Ca2 +结合蛋白质,GCAPs,细胞质的隔间。相比之下,没有公认的细胞外配体已确定,这些感官环化酶被认为是孤儿受体。GC-E已经检测到在这两个视杆细胞和视锥细胞,而GC-F似乎只有在视杆细胞。破坏老鼠的基因编码GC-E导致锥的选择性变性,但不棒。特定的突变基因与人类GC-E两个先天性视网膜疾病(促进et al ., 1999)。
类似的病理观察结果在CNG 3-deficient老鼠(比尔et al ., 1999))。这些老鼠显示锥体细胞的损失和不良反应,模拟全色盲(比尔et al ., 1999))。CNG通道,另一个重要组成部分phototransduction机制,cGMP的主要分子目标在视杆细胞和视锥细胞的质膜。像视网膜guanylyl环化酶,两种截然不同,但同源,heterotetrameric CNG通道存在于哺乳动物的视网膜。CNG 3,高的Ca2 +导,在锥和CNG 1,较低的Ca2 +电导,棒。因此,在锥体光感受器,Ca2 +在黑暗的阶段是在两倍棒涌入(总离子电流的10%)。这些内部Ca2 +电流是由外部Ca比例中和2 +电流由Na+/ Ca2 +K+换热器在感光细胞的质膜和提供分子依据光响应时间的差异在锥和棒(弗林斯,1997)。
最后一个组件的cGMP-related phototransduction机械PDE6,酶降解cGMP光感受器。这种酶底物是cGMP的首选。酶是由一个合作机制,最大限度地激活包括激活transducin PDE6γ-subunit, cGMP的绑定到特定的变构在PDE6网站。在视网膜棒,PDE6存在于膜蛋白复合物。在活动状态,这是由复杂的形成(α-和β-subunits),和不活跃的状态由四聚体(α-、β-、γ2子单元)。第四单元,称为δ,作为监管PDE6复杂的组件和指导易位细胞质膜的酶活性通过绑定prenylated PDE6β-subunit糖基(Linari et al ., 1999)。棒的PDE6复杂膜光盘是与视紫红质和transducin上游两种蛋白质在phototransduction PDE6。视紫红质包含一个发色团(11 -顺式视网膜)授予对光子的能力。Transducin是heterotrimeric GTP-binding蛋白质(αβγ-trimer);它释放出一个活化络合物形成α-subunit和三磷酸鸟苷的分子相互作用与激活视紫红质。
Ca2 +绑定蛋白在phototransduction扮演重要角色(图6)。Recoverin结合Ca2 +这个复杂的抑制视紫红质激酶,它允许激活受光的视紫红质。钙调蛋白激活钙2 +结合的β-subunit视网膜CNG渠道和降低cGMP的亲和力。最后,guanylyl酸环化酶激活蛋白家族联盟(GCAP 1 - 3”)激活视网膜guanylyl环化酶在低(Ca2 +]我(促进et al ., 1999)。一般来说,所有这些Ca2 +结合蛋白质(Ca2 +]我传感器来促进一个集成的响应。在低(Ca2 +]我他们抑制cGMP的水解,刺激cGMP的合成,并提高cGMP打开天然气通道的能力,从而提高Ca2 +]我。相比之下,在高(Ca2 +]我他们有助于减少(cGMP)我和降低(Ca2 +]我。

cGMP的角色在脊椎动物phototransduction棒。光(高压)激活序列,视紫红质(Rh) transducin (TR)和6型磷酸二酯酶(PDE),导致水解cGMP的GMP。减少(cGMP)我导致天然气通道的关闭,CNG1,诱导超极化,一个关键的信号将光子能量转化为中枢神经系统的冲动。此外,关闭CNG1减少Ca的涌入2 +当Ca2 +射流持续通过阳离子交换器,减少(Ca2 +]我,它允许调制响应的光通过减少与(1)GCAPs阳离子的相互作用,导致视网膜的激活guanylyl环化酶(RGC)补充(cGMP)我;(2)钙调蛋白(CAM),导致其从CNG1离解,减少cGMP的力量打开通道;和(3)recoverin (Rec)抑制蛋白,防止与视紫红质激酶(RK)交互,这磷酸化视紫红质,使其失去活性。在黑暗中状态,transducin (TR)和磷酸二酯酶(PDE)是不活跃的,反映了低可用性的激活视紫红质(Rh)和(cGMP)我积累,保持开放的阳离子通道CNG1构象和感光细胞去极化的状态。此外,CNG1 open状态的提升(Ca2 +]我(1)GCAPs结合,抑制视网膜guanylyl环化酶(RGC);(2)钙调蛋白(CAM),导致其与CNG1协会,增加cGMP的力量来维护通道的打开状态;和(3)recoverin (Rec),促进视紫红质相互作用和抑制激酶(RK),其余视紫红质激活。图改编自目前在神经生物学的观点小普EN,卷。9日,尼克诺夫和羊肉道明。脊椎动物的感光光适应的分子机制、410 - 418年,1999年版权许可爱思唯尔的科学。
哺乳动物phototransduction机械结合的分子组件创建一个复杂的级联cGMP和Ca2 +严格将交互的外部能量(光光子的形式)内部消息(电脉冲)(无花果。6)。因此,在黑暗的状态,高水平的cGMP确保(Ca2 +]我约500海里和维护外节杆处于去极化的状态。在这些条件下,PDE6复杂弱活跃,CNG1通道是开放的,和质膜Na的活动+/ Ca2 +K+(Ca服务器限制的上升2 +]我。光触发的顺序激活视紫红质,transducin PDE6复杂,导致cGMP的水解。随后的减少(cGMP)我关闭CNG1通道和扰乱了Ca的通量2 +。(Ca2 +]我从而降低了约50纳米,与感光膜超极化有关。光致下降(Ca2 +]我由Ca感觉到2 +结合蛋白质,从而抑制活化的视紫红质,刺激guanylyl环化酶,从而增加(cGMP)我从光致激发调停复苏。
四。结论
在过去的十年里,随着分子克隆技术的出现,我们理解的家庭guanylyl环化酶在生理和病理生理过程及其作用显著扩大。然而,有许多突出的问题和令人兴奋的挑战未来。因此,精确的分子机制调节各个域之间的信息流动guanylyl环化酶调解receptor-effector耦合需要说明。调节细胞组件guanylyl环化酶活动,包括激酶和磷酸酶,定义很重要。guanylyl酸环化酶活性和细胞代谢之间的关系值得关注,特别是因为包括由ATP严格监管,改变细胞内浓度的核苷酸可能对热解色谱信号产生深远的影响。的集成guanylyl环化酶到一般机制之间的细胞信号转导和相声guanylyl环化酶系统和其他信号级联应该检查。最后,识别和表征的新受体下游cGMP将进一步定义了这个重要的细胞信号机制的生理作用。
确认
这项工作是支持由美国国立卫生研究院(HL59214, CA75123 CA79663)。K.A.L.由国立卫生研究院培训支持格兰特5 t32 CA09137。I.R.-S。被少数国家卫生研究院补充支持。摩根大通(J.P.由国家卫生研究院培训支持格兰特T32 DK07705。S.A.W我年代the Samuel M.V. Hamilton Professor of Medicine, Jefferson Medical College, Thomas Jefferson University, Philadelphia, PA.
脚注
↵1通信地址:斯科特·a·沃尔德曼医生说,医学博士,博士,铸造,临床药理学,托马斯杰弗逊大学,132年南十圣。1170主,19107年费城,宾夕法尼亚州。电子邮件:scott.waldman在}{mail.tju.edu
缩写
- 没有
- 一氧化氮
- (Ca2 +]我
- 细胞内钙
- 安培
- 腺苷′-O——(3-thiomonophosphate)
- ANP
- 心房利钠肽
- ANPCR
- ANP间隙受体
- ATPγS
- 腺苷′-O——(3-thiotriphosphate)
- 法国巴黎
- 脑利钠肽
- 雌性生殖道
- 囊性纤维化跨膜电导调节
- (cGMP)我
- 细胞内cGMP
- 天然气管道
- 循环nucleotide-gated通道
- 中国出版集团
- c型利钠肽
- 有限公司
- 一氧化碳
- 表皮生长因子受体
- 表皮生长因子受体
- 电子商务50
- 配体的浓度产生半峰的响应
- 以挪士
- 内皮细胞一氧化氮合酶
- GC
- guanylyl环化酶
- GCAP
- guanylyl环化酶激活蛋白
- PKA
- cAMP-dependent蛋白激酶
- 包裹
- cGMP-dependent蛋白激酶
- G蛋白质
- heterotrimeric G蛋白质
- 销售税
- 谷胱甘肽年代转移酶
- K d
- 配体的浓度产生半峰绑定
- KHD
- 激酶同源域
- K 我
- 配体的浓度产生半峰抑制
- K 米
- 底物的浓度产生半峰速度
- 聚合酶链反应
- 聚合酶链反应
- PDE
- 磷酸二酯酶
- 包括
- 微粒guanylyl环化酶
- PKC
- 蛋白激酶C
- PMA
- 佛波醇12-myristate 13-acetate
- PPIX
- 原卟啉IX
- 页面
- 聚丙烯酰胺凝胶电泳
- 国网公司
- 可溶性guanylyl环化酶
- SMC
- 平滑肌细胞
- 单核苷酸多态性
- 硝普酸钠
- 圣
- 热稳定肠毒素
- V 马克斯
- 最大酶速度
- 号
- 没有合酶
- HCN
- hyperpolarization-activated循环nucleotide-gated
- 美国社会的药理学和实验性疗法
引用
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Molecular mechanisms underlying vascular smooth muscle relaxation mediated by cyclic GMP. Cyclic GMP induces smooth muscle relaxation by reducing [Ca2+]i and desensitizing the contractile apparatus to Ca2+. Cyclic GMP reduces [Ca2+]i by (1) inhibiting Ca2+ influx through L-type Ca2+ channels; (2) increasing Ca2+ efflux through activation of (2d) the Ca2+-pumping ATPase and (2b) the Na+/Ca2+ exchanger; also, cGMP may produce membrane hyperpolarization through activation of (2c) the Na+/K+ ATPase and (2a) K+ channels, thereby increasing Ca2+ extrusion by the Na+/Ca2+ exchanger; (3) increasing of Ca2+ sequestration through activation of the sarcoplasmic reticulum Ca2+-pumping ATPase [Ph, phospholamban]; and (4) decreasing of Ca2+ mobilization through inhibition of agonist-induced IP3 formation or inhibition of the IP3 receptor in the sarcoplasmic reticulum. R, receptor; G, G protein; PLC, phospholipase C; IP3R, IP3 receptor. Cyclic GMP desensitizes the contractile apparatus to Ca2+ (5) probably by activating myosin light chain phosphatase, resulting in dephosphorylation of the 20 kDa myosin light chain.](https://pharmrev.aspetjournals.org/content/pharmrev/52/3/375/F4.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![Regulation of intestinal secretion by the ST and GC-C. Bacteria, such as E. coli, containing plasmids encoding a member of the homologous peptide family of STs colonize the intestine after the consumption of contaminated food and/or water. Bacterial colonization leads to production of ST in the gut lumen, which specifically recognizes and binds to the extracellular domain of GC-C, expressed in the brush border membranes of intestinal mucosa cells from the duodenum to the rectum. Interaction of ST and the extracellular domain of GC-C is translated across the plasma membrane into activation of the cytoplasmic catalytic domain resulting in the production and accumulation of [cGMP]i. This cyclic nucleotide binds to and activates PKG II, also localized in the intestinal cell brush border membrane. Also, cGMP may activate PKA, either directly or by inhibiting a cAMP-specific PDE and inducing the accumulation of cAMP. The CFTR that is colocalized with GC-C and PKG II in brush border membranes is a substrate for that protein kinase and PKA. CFTR is a chloride channel, and its phosphorylation by PKA or PKG results in a persistent open state, permitting chloride to flow down its concentration gradient from the intracellular to the extracellular compartment. Other ion channels and transporters in the cell maintain the electroneutrality of ST-induced chloride efflux. Vectoral water flux from the basolateral to the apical surface is driven by these ionic conductances, resulting in the accumulation of fluid and electrolytes in the intestinal lumen and secretory diarrhea.](https://pharmrev.aspetjournals.org/content/pharmrev/52/3/375/F5.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![The role of cGMP in phototransduction in vertebrate rods. Light (hv) activates, in sequence, rhodopsin (Rh), transducin (TR), and phosphodiesterase type 6 (PDE), resulting in the hydrolysis of cGMP to GMP. Decreased [cGMP]iresults in the closure of the CNG channel, CNG1, inducing hyperpolarization, a key signal translating photon energy into central nervous system impulses. In addition, closure of CNG1 reduces the influx of Ca2+ while Ca2+ efflux persists through the cation exchanger, reducing [Ca2+]i, which permits modulation of the response to light by decreasing interaction of that cation with (1) GCAPs, resulting in the activation of retinal guanylyl cyclases (RGC) to replenish [cGMP]i; (2) calmodulin (CAM), resulting in its dissociation from CNG1, reducing the potency of cGMP to open that channel; and (3) recoverin (Rec), which inhibits that protein, preventing interaction with rhodopsin kinase (RK), which phosphorylates and inactivates rhodopsin. In the dark state, transducin (TR) and the phosphodiesterase (PDE) are inactive, reflecting the low availability of activated rhodopsin (Rh), and [cGMP]i accumulates, maintaining the cation channel CNG1 in the open conformation and photoreceptors in a depolarized state. In addition, CNG1 in the open state elevates [Ca2+]i, which binds to (1) GCAPs, inhibiting retinal guanylyl cyclase (RGC); (2) calmodulin (CAM), resulting in its association with CNG1, increasing the potency of cGMP to maintain that channel in the open state; and (3) recoverin (Rec), promoting interaction with and inhibition of rhodopsin kinase (RK), potentiating rhodopsin activation. Figure adapted from Current Opinion in Neurobiology, vol. 9, Pugh EN Jr, Nikonov S and Lamb TD. Molecular mechanisms of vertebrate photoreceptor light adaption, 410–418, Copyright 1999, with permission from Elsevier Science.](https://pharmrev.aspetjournals.org/content/pharmrev/52/3/375/F6.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}