条文本

摘要
肺动脉高压的发展是慢性阻塞性肺疾病(COPD)患者预后不良的标志,影响死亡率和生活质量。尽管COPD的肺动脉高压传统上被认为是血管床的肺气肿性破坏和/或缺氧的结果,但最近的研究表明,这些因素都与肺动脉压没有很好的相关性。新的人体和动物实验数据开始表明,在这种情况下,肺动脉高压可能是烟草烟雾直接影响肺内血管的结果,导致控制血管收缩、血管扩张和血管细胞增殖的介质产生异常,最终导致血管重塑异常和血管生理异常。这些变化在许多方面与其他形式的肺动脉高压相似,这表明用于原发性肺动脉高压的治疗可能对COPD患者有益。
- eNOS,内皮型一氧化氮合酶
- ET-1, endothelin-1
- FEV1, 1秒内用力呼气量
- NO,一氧化氮
- 巴勒斯坦权力机构o2动脉氧张力
- VEGF,血管内皮生长因子
- 慢性阻塞性肺疾病
- 肺动脉高压
- 香烟烟雾
- 血管活性的介质
- 肺气肿
数据来自Altmetric.com
慢性阻塞性肺疾病(COPD)已成为发病率和死亡率的第四大原因,在美国造成50万潜在寿命损失,直接成本超过180亿美元,在英国约14亿美元。1在慢性阻塞性肺病患者中,临床诊断的肺心病一直被认为是一个重要的负面预后标志,即使在1秒用力呼气量(FEV)校正后也是如此1).2当测量肺血流动力学时,静息时平均肺动脉压的升高也被证明是一个重要的预后变量。2平均压力每增加10毫米汞柱,死亡率增加4倍以上。3.
COPD中肺动脉高压的实际发病率尚不清楚,尽管早期研究估计每年有6%的COPD患者会发生肺心病。最近的一项研究检查了伴有FEV的COPD患者1在大约6年的时间间隔内,从预测的<35%(28.2%的受试者)到预测的> - 50%(26.5%的受试者),没有人在休息时患有肺动脉高压。4在第二次评估中发现,平均肺动脉压缓慢上升(每年0.28 mm Hg), 25%的患者现在静息肺动脉压为>20 mm Hg,这在第一次评估时运动时出现肺动脉高压的受试者中更为常见。这一进展速度与早期研究中发现的0.6 mm Hg的平均增长速度相似。5对这些数据的推断表明,相当数量的COPD患者在病程中会出现肺动脉高压,并因此增加发病率和死亡率。
值得注意的是,在COPD患者中观察到的肺动脉压升高通常是适度的。基于对特发性(“原发性”)肺动脉高压患者群体的数据分析,COPD患者的心肺血流动力学变化本身并不会对死亡率产生强烈影响。因此,COPD和肺动脉高压患者死亡率明显增加的事实表明,肺部正在发生一系列复杂的相互作用。
在特发性肺动脉高压和继发原因(如结缔组织疾病)相关的肺动脉高压患者中,除了对死亡率的影响外,高血压对功能能力、运动不耐受和健康相关的生活质量也有广泛的影响。虽然通气功能异常和通气储备不足是限制COPD患者峰值运动能力的常见重要因素,但影响特发性肺动脉高压患者整体生活质量的机制可能也适用于COPD和肺动脉高压患者。
这篇论文并不是一个全面的综述,而是一个简短的回顾,提出了各种理论来解释肺血管功能障碍的病因和吸烟者肺动脉高压的发展。大多数关于COPD肺动脉高压的信息来自于人体研究,相对较少的实验动物模型研究了暴露于烟雾对肺血管的影响。然而,有研究表明,长期暴露在外交系哈特利品系豚鼠的烟雾中与肺动脉压升高有关,而且这种情况只发生在人口的一个子集中,6与人类的情况类似。下面我们将更详细地考虑实验研究。
吸烟者肺血管的结构变化
吸烟者的肌性肺动脉腔室的结构已在尸检和手术切除标本中得到广泛检查。然而,通常很难使用这些报告来推断病理生理机制。虽然许多研究检查了COPD受试者的病理和/或生理变化,但其他研究使用有无肺气肿作为鉴别标准,而其他研究则关注吸烟者与戒烟者或不吸烟者之间的差异。
对中重度COPD患者血管系统的研究一致发现内膜发生了变化,7,8,9,10伴有局灶性纤维弹性增厚和纵向肌肉增加。9内膜厚度的增加已被证明是由平滑肌细胞增殖和弹性蛋白增加以及胶原沉积引起的。10
在非常严重的肺气肿患者中,外径为100-200 μm的血管的总壁厚(包括内膜和肌中膜)已被证明与运动时的肺动脉压或休息时和运动时肺动脉压的差有关。11这表明这种类型的重塑会导致肺血管扩张能力的降低。然而,关于肺动脉壁肌肉的数量,有相互矛盾的数据。许多研究未能发现任何异常或仅发现壁厚的微小增加,即使在严重COPD患者的肺中也是如此。7,10其他人发现,与不吸烟者相比,吸烟者的血管内侧增厚,12从非吸烟者到无气流阻塞的吸烟者,再到有气流阻塞的吸烟者,除了肌壁厚度有明显的增加外,还伴随着较小尺寸血管比例的增加。8然而,肌肉内侧增厚,相对于整体血管壁增厚,似乎与肺动脉高压的严重程度或血管对氧气的反应能力没有任何明确的关系。9
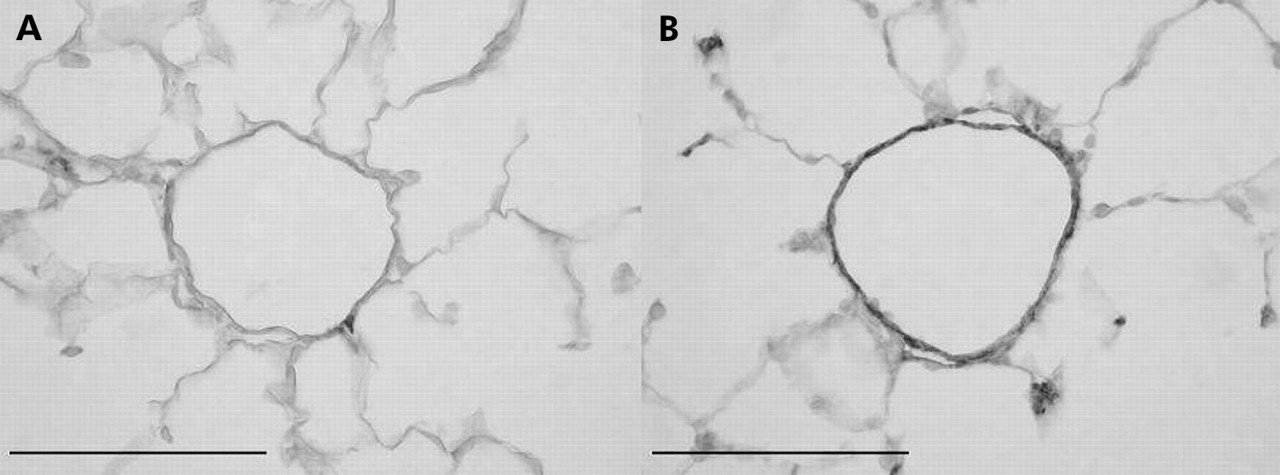
在严重COPD患者中,可一致发现小动脉(通常肌肉化较差)肌肉化,包括圆形和纵向的肌肉外衣,通常伴有纵向管的形成,后者表明异常重塑。暴露于香烟烟雾的动物模型清楚地显示,毗邻肺泡导管的部分肌肉化动脉的肌肉化增加(图1),6,13这些研究表明,这种情况不会在戒烟后恢复。
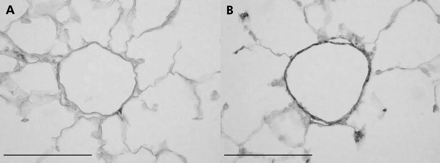
在暴露于室内空气(A)或香烟烟雾(B) 6个月的雌性豚鼠中,平滑肌动蛋白染色显示毗邻肺泡管的小的、正常部分肌肉化的动脉的肌肉化。来自对照动物(A)的血管只有几块平滑肌,而来自暴露在烟雾中的动物(B)的血管几乎完全肌肉化。在慢性阻塞性肺病患者的肺部也有类似的变化。放大棒= 100 μm。
很少有研究检查肌肉动脉的外膜室,没有发现一致的变化。7,9,10然而,一个例外是,吸烟者的动脉,无论是否患有COPD,似乎有更多的外源性CD8淋巴细胞。10这些发现表明,血管结构重组——肌肉化和部分肌肉化的小动脉的肌肉化增加——可能是吸烟的直接后果,在人类中,这种反应存在相当大的变异性,这种反应可能先于COPD的血管生理改变。然而,这些报告的含义,总的来说,肌肉化增加本身并不能为COPD患者肺动脉高压的存在提供一致的解释。
吸烟诱发肺动脉高压的病因研究
缺氧和肺气肿
早期文献提示肺气肿破坏肺血管床导致肺动脉高压。然而,已发表的研究未能显示肺动脉压与肺气肿之间或肺气肿与右心重量之间的直接相关性,以支持这一说法。14日,15在动物模型中也发现了这种相关性的缺乏,在这些动物模型中,长期暴露于烟雾中会产生肺气肿性气隙扩大,毛细血管密度降低,毛细血管狭窄,但这些变化与肺动脉压升高没有关系。
缺氧,如在高海拔地区,肯定会引起肺动脉高压。尽管在严重肺气肿患者中经常同时存在肺动脉高压和低氧血症,但显然,除了严重缺氧状态外,缺氧本身并不是血管重组、肺动脉高压和动脉氧紧张的主要驱动因素(Pao2)尚未被确定为平均肺动脉压的独立预测因子。14此外,尽管人们普遍认为肺气肿中存在肺动脉高压反映了肺气肿继发的毛细血管床的丧失和/或缺氧,但肺动脉高压实际上似乎与缺氧无关,因为在没有严重低氧血症的患者中(Pao2>7.3 kPa),平均肺动脉压被认为是最显著的预后因素。在需要长期氧疗的严重气流阻塞患者中,平均肺动脉压超过25毫米汞柱使5年生存率降低到36.3%,而当肺动脉压小于25毫米汞柱时,5年生存率为62.2%。16此外,一项临床试验评估了在轻度至中度低氧血症的情况下,对无至轻度肺动脉高压患者的氧给药,以及睡眠期间的进一步去饱和,在2年的随访期内,肺血流动力学没有改变。最后,对严重低氧血症的受试者夜间吸氧确实降低了肺动脉压,但对这些患者肺部的病理分析显示,不同患者的血管异常是一致的,因此不能解释对运动的不同生理反应,也不能解释某些患者对氧气的血管反应。9
肺血管的动态变化
另一种模式认为肺动脉高压是肺血管床动态变化的结果。这包括两个并不相互排斥的一般假设。“结构”理论提出,血管的早期形态变化确实发生,但最初在流量增加时不干扰血管扩张。然而,当结构改变到血管不顺应时,流量增加导致肺动脉压增加。7为了支持这一假设,在大鼠模型中报道了慢性烟雾暴露后肺动脉顺应性的下降。17另外,也有人认为血管的顺应性可能受到肺功能变化的影响,气体滞留导致血管受压。7,18事实上,肺动脉压似乎与气流阻塞的程度相关,即使在轻度气流受限的患者中也是如此。7然而,在肺减容手术(一种通过气体捕获减少血管压迫的技术)后测量血管动力学时,无论在休息时还是在运动时,肺动脉压力都没有变化,这表明要么非常少量的气体捕获有主要影响,要么在这种严重疾病的受试者中,由于结构改变,血管已经僵硬,气体捕获不会产生附加效应。然而,这一概念已经得到了唯一的动物研究的支持;6在豚鼠中,气流阻塞的生理证据伴随着肺动脉压的升高,而具有类似程度的肺气肿但只有轻微气流改变的动物肺动脉压没有明显升高。
内皮功能障碍与血管重构
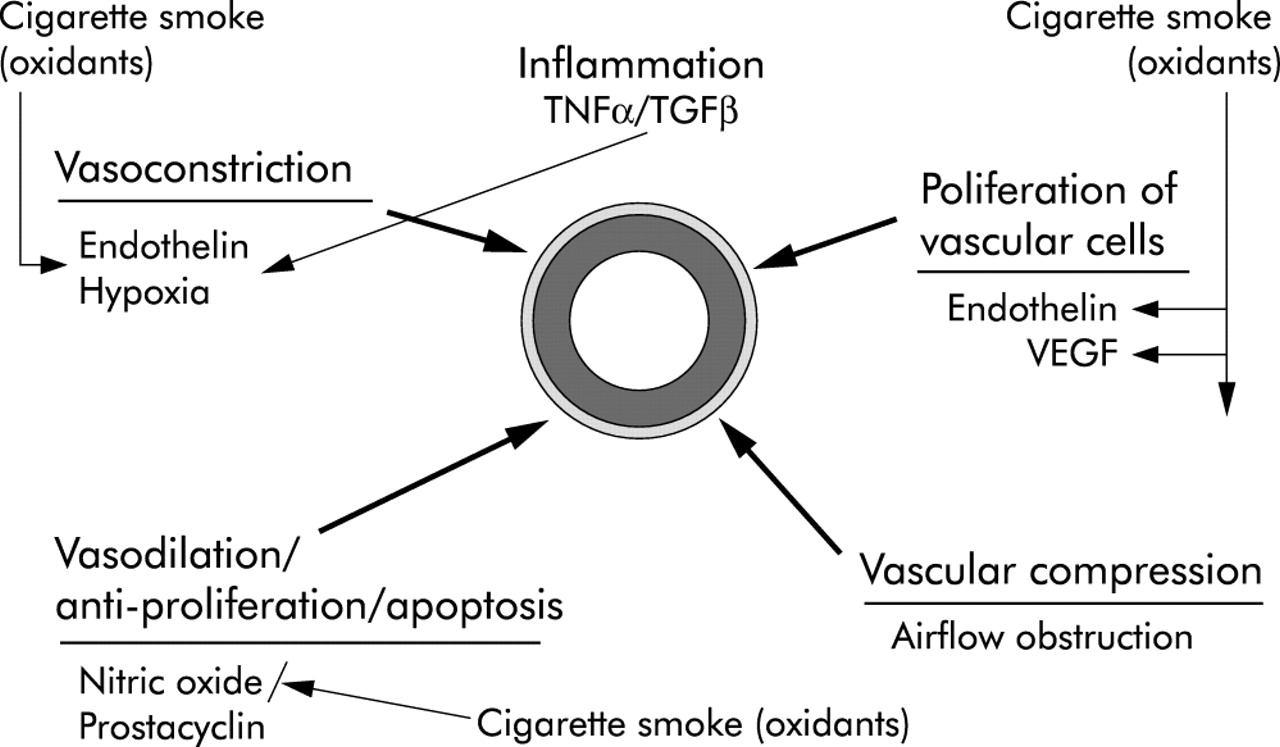
在大多数形式的非copd相关肺动脉高压中,引起血管重构的初始刺激的性质尚不清楚,尽管经常假定内皮细胞损伤。“内皮功能障碍”是内皮细胞结构和/或介质产生的各种异常的总称。内皮功能障碍理论19认为张力和/或细胞复制介质的产生或响应存在调节障碍,从而最终导致血管重塑和血管生理异常(图2)。由于这些介质大多数影响平滑肌生长和增殖,内皮功能障碍被认为是导致肺动脉高压患者血管肥大和血管结构异常的一种力量。此外,内皮损伤可能导致内皮屏障的丧失,使循环介质和生长因子直接影响底层的血管壁。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
图中显示了与香烟烟雾相关的各种过程,这些过程导致小肺动脉壁上的血管收缩、血管舒张和细胞增殖。所显示的大多数影响可能是由烟雾中的氧化剂作用于血管壁直接介导的。血管显示在中心,肌肉层和外延层突出显示。烟雾中的氧化剂会导致血管收缩剂内皮素、血管增殖剂内皮素和血管内皮生长因子(VEGF)的产生。烟雾通过降低一氧化氮生物活性而干扰一氧化氮介导的血管舒张。作为慢性气流阻塞的一部分,空气滞留似乎在早期通过诱导血管压迫发挥作用,而缺氧导致血管收缩,但仅在氧张力极低时才会发生。
在吸烟者中,香烟烟雾对血管的直接作用可能导致内皮功能障碍和重构,从而改变细胞因子或血管活性介质的平衡。13它也可能是由血管壁的炎症反应引起的。10这种功能障碍已在COPD患者的肺外肺动脉中发现,其中有一氧化氮依赖性松弛的丧失。20.这种效应不是由于异常的膜受体或l-精氨酸缺乏,但似乎与内膜增厚有关。
有大量关于内源性介质(包括血管扩张剂,如一氧化氮(NO)和脂质介质前列环素,血管收缩剂,如内皮素,有丝分裂刺激剂血管内皮生长因子(VEGF)在其他病因的肺动脉高压中的作用的文献。21日,22
内皮源性一氧化氮通常被认为是一种血管扩张剂,并已被证明可以抑制平滑肌增殖以及下调血管内皮素的产生。23它是由内皮型一氧化氮合酶(eNOS)的活性产生的,剪切应力和增加的肺血流量似乎都能调节eNOS活性。在小鼠中,eNOS的过表达可以预防低氧性肺动脉高压,24而eNOS敲除小鼠仅暴露于轻度缺氧时会出现肺动脉高压。25
前列环素是一种血管扩张剂,也可以预防血管重塑和肺动脉高压。虽然前列环素过表达可以保护小鼠对抗缺氧引起的肺动脉高压,26前列环素受体缺乏的小鼠在缺氧环境中会出现严重的肺动脉高压。27在各种形式的肺动脉高血压患者的肺动脉中发现前列环素合成酶水平下降,静脉注射前列环素(epoprostenol)可以改善血流动力学和生存,同时降低循环内皮素和增加VEGF水平。28
内皮素-1 (ET-1)是一种强大的内皮来源的血管收缩肽。在实验性缺氧和野花碱诱导的肺动脉高压以及肺动脉高压患者中,内皮素水平升高,而ET-1水平与此类患者的肺血管阻力相关。29ET-1受体拮抗剂在原发性肺动脉高压患者中产生血流动力学和临床改善。30.
尽管肺动脉高压的内皮细胞产生的免疫反应性VEGF增加,但VEGF在肺动脉高压中的作用尚不完全清楚。在大鼠中,化学VEGF受体阻断会产生严重的肺动脉高压31通过病毒载体传递VEGF可预防低氧性肺动脉高压。32在正常的主动脉内皮细胞中,VEGF诱导前列环素和NO的产生,33因此有利于血管舒张。然而,有研究表明,在血管损伤的情况下,VEGF通过作为一种生长因子来增强损伤。VEGF也诱导基质金属蛋白酶-1,34一种可能有助于血管重塑的效应(见下文)。
很少有研究在人类吸烟者和COPD患者中检测这些介质,报道的结果也不完全一致。基于免疫组化方法的报告发现,吸烟者肺内动脉eNOS染色明显减少,大量肺样本中eNOS蛋白也减少。10在少数COPD继发肺动脉高压患者的肺血管中发现内皮素免疫反应性增加。29此外,在吸烟者和COPD患者中发现循环内皮素水平升高。35有趣的是,在没有COPD的吸烟者和中度COPD的吸烟者中,动脉免疫反应染色、肺体积蛋白和VEGF的mRNA水平升高,但在严重肺气肿患者的肺中下降。36没有关于吸烟者前列环素水平的数据。
许多研究已经在实验系统中观察了这些介质的产生。在内皮细胞培养模型中,香烟烟雾诱导内皮素的产生,降低eNOS的产生和活性。37使用大体积肺或更敏感的显微解剖方法,可以对小肺内动脉的基因表达水平进行特定的评估,在只暴露一次烟雾的动物中,发现内皮素和VEGF mRNA水平和免疫反应蛋白迅速升高。38这种上调在长期暴露的情况下持续了6个月13(莱特J等,未发表的数据)。与上述人类数据相比,这些研究中eNOS mRNA和免疫反应蛋白也有所增加。因此,虽然人和动物的VEGF和内皮素数据相对一致,但eNOS数据存在差异。
尽管如此,这些发现有力地表明,香烟烟雾直接诱导在动脉管壁内产生控制动态血管收缩和血管扩张的血管活性介质。虽然这些影响在近亲繁殖的动物品系中有些刻板,因为每种动物都表现出相当相似的反应,但在近亲繁殖的哈特利品系豚鼠中,只有一些豚鼠在长期暴露于香烟烟雾后会出现肺动脉高压,这些介质的基因表达水平有明显的变化,13在暴露于烟雾6个月的豚鼠中,介质产生的水平与肺动脉压力直接相关(Wright J等,未发表的数据)。这一观察结果再次表明,吸烟对血管系统的内在影响是血管重塑和肺动脉高压的直接驱动因素,同时也提出了一种可能性,即控制这些介质表达水平的遗传多态性可能在决定哪些人类吸烟者患上肺动脉高压方面具有重要意义。
细胞增殖和血管重塑
尚未在人类吸烟者的肺血管系统中测量到细胞增殖。烟雾暴露3个月后,大鼠肺动脉(直径约450 μm)成纤维细胞、胶原束、弹性层体积比例增加,平滑肌细胞减少。17在与细支气管相邻的较小肌肉血管和与肺泡管相邻的部分肌肉化的小动脉中,急性体内烟雾暴露与细胞增殖有关,并且这种增殖在持续几个月的慢性烟雾暴露中保持。13在急性烟雾暴露的肺片外植体模型中,内皮素拮抗剂、内皮素转换酶抑制剂和谷胱甘肽可显著降低细胞增殖,后者表明氧化剂在诱导细胞增殖中具有潜在作用。这些发现再次支持了烟雾对血管重塑的直接影响,这种影响不仅与气流阻塞无关,而且与烟雾引起的炎症无关。
蛋白酶和血管重塑
据我们所知,还没有研究证实蛋白酶参与了人类COPD中的血管重塑,但各种(非吸烟)肺动脉高压的动物模型已经清楚地证明蛋白酶在促进血管重塑方面的作用。21暴露于缺氧或单野百合碱会导致大鼠肺动脉中内源性丝氨酸弹性蛋白酶的表达,21在这些模型中,弹性蛋白酶抑制可以防止肺动脉高压以及血管结构的改变。内源性丝氨酸弹性蛋白酶的作用被认为是激活多种基质金属蛋白酶,这些蛋白酶本身可能引起血管重构以及从基质中释放生长因子导致细胞增殖。丝氨酸和金属弹性蛋白酶也诱导腱蛋白c的表达,21一种引起生长因子受体磷酸化并增强对其他生长因子反应的物质。这些研究表明丝氨酸蛋白酶和金属蛋白酶在血管重塑过程中都是不可或缺的。
一个单一的动物模型研究了口服丝氨酸弹性蛋白酶抑制剂对暴露在香烟烟雾中的动物血管结构的影响,发现小血管细胞增殖和肌肉化的数量显著减少。39我们在这些动物的肺内小动脉中观察到各种基质金属蛋白酶的基因表达上调(Wright J, Churg A,未发表数据)。
治疗的意义
值得注意的是,尽管有大量证据反对COPD的肺动脉高压仅仅是由于缺氧和/或肺气肿破坏肺血管床的观点,但从分类和治疗的角度来看,COPD相关的肺动脉高压往往与其他主要通气条件下的肺动脉高压混为一谈。事实上,肺动脉高压的分类是在世界卫生组织国际会议上提出的,最近在2003年威尼斯举行的第三届世界国际会议上进行了更新,22将COPD中的肺动脉高压置于“低氧血症相关的肺动脉高压”标题下,这一分组包括COPD、间质性肺病、睡眠呼吸障碍、肺泡低通气障碍和慢性高海拔暴露。40不幸的是,这种普遍有用的分类没有考虑到上面概述的大量证据,这些证据表明肺动脉高压可能不仅仅与COPD的缺氧有关,或者新的机制数据表明,香烟烟雾通过上调介质导致血管结构重构和血管功能的动态生理变化对肺血管有直接影响。
认识到这些直接由烟雾引起的血管病理生理过程应该导致更积极地追求和研究针对其废除或改善的具体治疗方法。这可能会降低吸烟者和COPD患者肺动脉高压的后果,不仅是在死亡率方面,而且还可以提高功能能力、运动耐受性和健康相关的生活质量。尽管目前可用的肺动脉高压特异性治疗如前列腺素、内皮素受体拮抗剂和磷酸二酯酶抑制剂价格昂贵,但它们实际上可能为COPD患者提供重要的有益影响,并导致医疗保健系统的总体成本降低。
综上所述,虽然肺血管受到气流阻塞的影响,并可能受到严重的肺气肿和缺氧的影响,但传统的认为肺气肿和缺氧可以解释COPD中存在肺动脉高压的观点并没有真正得到数据的支持。有证据表明,在患有严重的在缺氧时,夜间吸氧是有益的,但这种方法对大多数COPD和肺动脉高压患者没有价值。现在是时候基于新的病理生理学见解,重新审视COPD肺动脉高压的影响和潜在治疗。
参考文献
脚注
这项研究得到了加拿大卫生研究院62693加元的资助。这项工作独立于资助者。
利益竞争:JLW和AC已获得阿斯利康(AstraZeneca)的资金,用于研究肺气肿的动物模型,并测试正在开发的各种化合物的有效性。RDL在2002年(1000加元)、2003年(2500加元)和2004年(1200加元)担任Actelion的顾问委员会成员,担任葛兰素史克公司(2003年为3375加元)和北方治疗公司(2004年为1750加元)的顾问,并从Actelion收取持续健康教育活动讲座费用(2002年为2000加元;2003年为750加元;2004年为1250美元),2003 - 2004年为参与一项多中心临床试验提供了34 000加元的研究资助。